Expression of human‐specific ARHGAP11B in mice leads to neocortex expansion and increased memory flexibility
In this project led by the Huttner lab a mouse line in which we engineered the ancestral ARHGAP11A gene to mimic the human specific gene with a proposed essential role in the brain cortex expansion ARHGAP11B was shown to both increase brain size and cognitive abilities in mice.
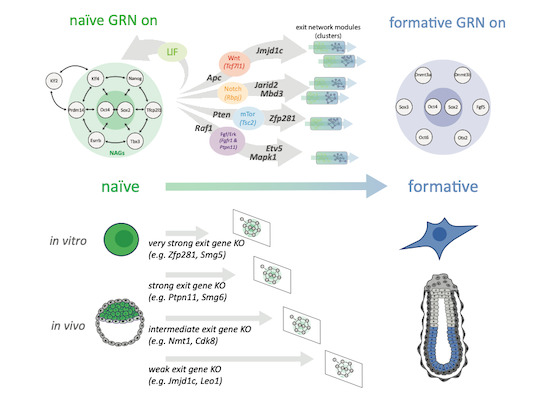
Cooperative genetic networks drive embryonic stem cell transition from naïve to formative pluripotency
A resource of of guide RNA construct that we built using high throughput recombineering was used in this large scale project combining CRISPR/Cas9 gene disruption in haploid embryonic stem cells, transcriptomics and systems biology analysis to delineate the regulatory circuits that control the transition to formative pluripotency from the naïve state of the epiblast cells - a cruical step in the early mammalian development.
We provided BAC-transgenes for this large scale study of DNA-repair. Through fluorescent tracking of the recruitment of DNA repair-related proteins expressed at near endogenous levels to sites of DNA damage and systems biology analysis this project provided a comprehensive and precise description of the dynamics of human DNA repair mechanisms.
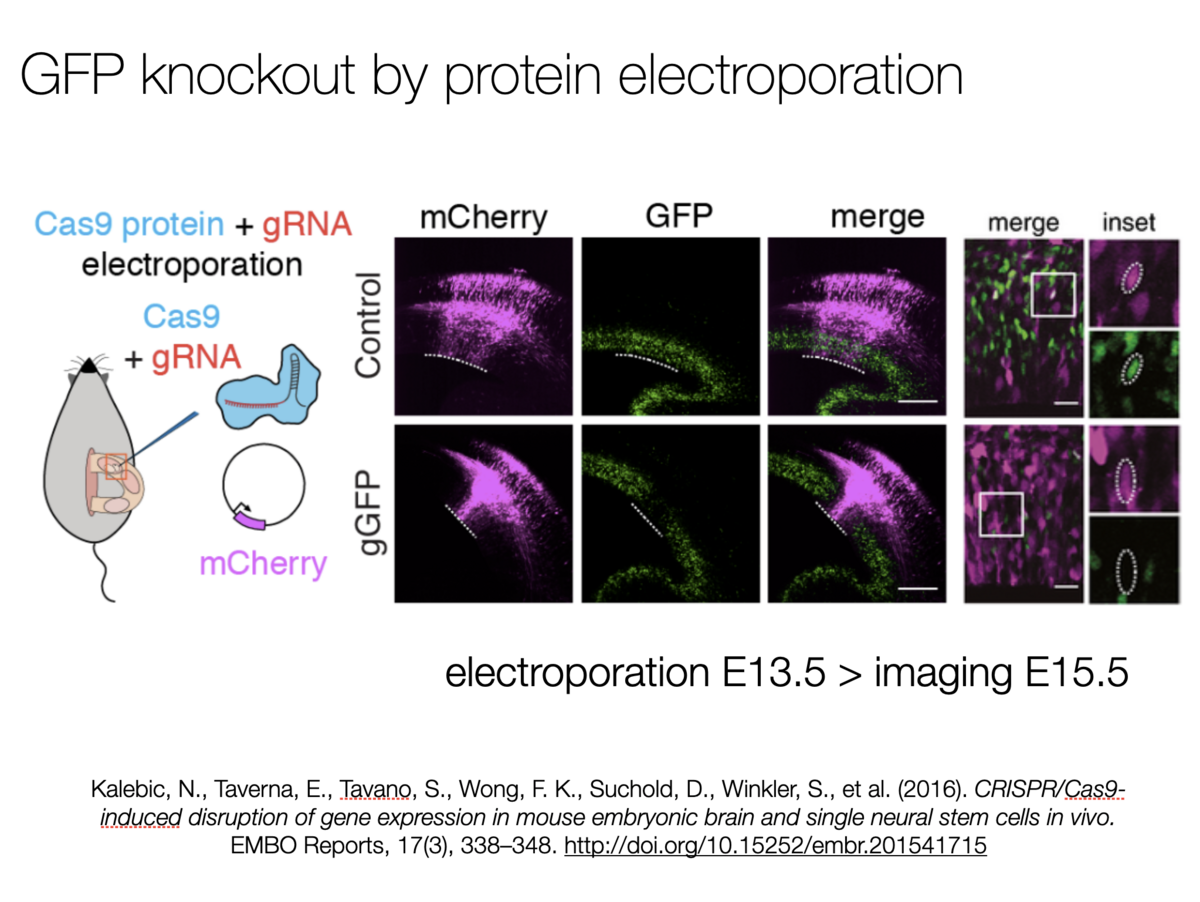
CRISPR/cas9-induced disruption of gene expression in mouse embryonic brain and single neural stem cells in vivo
This collaboration of the Huttner lab and GEF introduced use of CRISPR/Cas9 gene disruption in situ as an efficient and cost effective tool for function discovery in neuroscience.
Shamini Ramkumar Thirumalasetty, Tina Schubert, Ronald Naumann, Ilka Reichardt, Marie-Luise Rohm, Dana Landgraf, Florian Gembardt, M Peitzsch, Michaela F Hartmann, Mihail Sarov, Stefan A Wudy, Nicole Reisch, Angela Huebner, Katrin Koehler A Humanized and Viable Animal Model for Congenital Adrenal Hyperplasia-CYP21A2-R484Q Mutant Mouse. Int J Mol Sci, 25(10) Art. No. 5062 (2024)
Open Access DOI
Congenital Adrenal Hyperplasia (CAH) is an autosomal recessive disorder impairing cortisol synthesis due to reduced enzymatic activity. This leads to persistent adrenocortical overstimulation and the accumulation of precursors before the blocked enzymatic step. The predominant form of CAH arises from mutations in CYP21A2, causing 21-hydroxylase deficiency (21-OHD). Despite emerging treatment options for CAH, it is not always possible to physiologically replace cortisol levels and counteract hyperandrogenism. Moreover, there is a notable absence of an effective in vivo model for pre-clinical testing. In this work, we developed an animal model for CAH with the clinically relevant point mutation p.R484Q in the previously humanized CYP21A2 mouse strain. Mutant mice showed hyperplastic adrenals and exhibited reduced levels of corticosterone and 11-deoxycorticosterone and an increase in progesterone. Female mutants presented with higher aldosterone concentrations, but blood pressure remained similar between wildtype and mutant mice in both sexes. Male mutant mice have normal fertility with a typical testicular appearance, whereas female mutants are infertile, exhibit an abnormal ovarian structure, and remain in a consistent diestrus phase. Conclusively, we show that the animal model has the potential to contribute to testing new treatment options and to prevent comorbidities that result from hormone-related derangements and treatment-related side effects in CAH patients.
Paula Cubillos, Nora Ditzer, Annika Kolodziejczyk, Gustav Schwenk, Janine Hoffmann, Theresa M Schütze, Razvan Derihaci, Cahit Birdir, Johannes Em Köllner, Andreas Petzold, Mihail Sarov, Ulrich Martin, Katherine S. Long, Pauline Wimberger, Mareike Albert The growth factor EPIREGULIN promotes basal progenitor cell proliferation in the developing neocortex. EMBO J, 43(8) 1388-1419 (2024)
Open Access DOI
Neocortex expansion during evolution is linked to higher numbers of neurons, which are thought to result from increased proliferative capacity and neurogenic potential of basal progenitor cells during development. Here, we show that EREG, encoding the growth factor EPIREGULIN, is expressed in the human developing neocortex and in gorilla cerebral organoids, but not in the mouse neocortex. Addition of EPIREGULIN to the mouse neocortex increases proliferation of basal progenitor cells, whereas EREG ablation in human cortical organoids reduces proliferation in the subventricular zone. Treatment of cortical organoids with EPIREGULIN promotes a further increase in proliferation of gorilla but not of human basal progenitor cells. EPIREGULIN competes with the epidermal growth factor (EGF) to promote proliferation, and inhibition of the EGF receptor abrogates the EPIREGULIN-mediated increase in basal progenitor cells. Finally, we identify putative cis-regulatory elements that may contribute to the observed inter-species differences in EREG expression. Our findings suggest that species-specific regulation of EPIREGULIN expression may contribute to the increased neocortex size of primates by providing a tunable pro-proliferative signal to basal progenitor cells in the subventricular zone.
2022
Felipe Mora-Bermúdez, Philipp Kanis, Dominik Macak, Jula Peters, Ronald Naumann, Lei Xing, Mihail Sarov, Sylke Winkler, Christina Eugster Oegema, Christiane Haffner, Pauline Wimberger, Stephan Riesenberg, Tomislav Maricic, Wieland Huttner, Svante Pääbo Longer metaphase and fewer chromosome segregation errors in modern human than Neanderthal brain development. Sci Adv, 8(30) Art. No. eabn7702 (2022)
Open Access DOI
Since the ancestors of modern humans separated from those of Neanderthals, around 100 amino acid substitutions spread to essentially all modern humans. The biological significance of these changes is largely unknown. Here, we examine all six such amino acid substitutions in three proteins known to have key roles in kinetochore function and chromosome segregation and to be highly expressed in the stem cells of the developing neocortex. When we introduce these modern human-specific substitutions in mice, three substitutions in two of these proteins, KIF18a and KNL1, cause metaphase prolongation and fewer chromosome segregation errors in apical progenitors of the developing neocortex. Conversely, the ancestral substitutions cause shorter metaphase length and more chromosome segregation errors in human brain organoids, similar to what we find in chimpanzee organoids. These results imply that the fidelity of chromosome segregation during neocortex development improved in modern humans after their divergence from Neanderthals.
Aleksandra Spiegel, Chris Lauber, Mandy Bachmann, Anne-Kristin Heninger, Christian Klose, Kai Simons, Mihail Sarov, Mathias J. Gerl A set of gene knockouts as a resource for global lipidomic changes. Sci Rep, 12(1) Art. No. 10533 (2022)
Open Access DOI
Enzyme specificity in lipid metabolic pathways often remains unresolved at the lipid species level, which is needed to link lipidomic molecular phenotypes with their protein counterparts to construct functional pathway maps. We created lipidomic profiles of 23 gene knockouts in a proof-of-concept study based on a CRISPR/Cas9 knockout screen in mammalian cells. This results in a lipidomic resource across 24 lipid classes. We highlight lipid species phenotypes of multiple knockout cell lines compared to a control, created by targeting the human safe-harbor locus AAVS1 using up to 1228 lipid species and subspecies, charting lipid metabolism at the molecular level. Lipid species changes are found in all knockout cell lines, however, some are most apparent on the lipid class level (e.g., SGMS1 and CEPT1), while others are most apparent on the fatty acid level (e.g., DECR2 and ACOT7). We find lipidomic phenotypes to be reproducible across different clones of the same knockout and we observed similar phenotypes when two enzymes that catalyze subsequent steps of the long-chain fatty acid elongation cycle were targeted.
Tina Schubert, Nicole Reisch, Ronald Naumann, Ilka Reichardt, Dana Landgraf, Friederike Quitter, Shamini Ramkumar Thirumalasetty, Anne-Kristin Heninger, Mihail Sarov, M Peitzsch, Angela Huebner, Katrin Koehler CYP21A2 Gene Expression in a Humanized 21-Hydroxylase Mouse Model Does Not Affect Adrenocortical Morphology and Function. J Endocr Soc, 6(6) Art. No. bvac062 (2022)
Open Access DOI
Steroid 21-hydroxylase is an enzyme of the steroid pathway that is involved in the biosynthesis of cortisol and aldosterone by hydroxylation of 17α-hydroxyprogesterone and progesterone at the C21 position. Mutations in CYP21A2, the gene encoding 21-hydroxylase, cause the most frequent form of the autosomal recessive disorder congenital adrenal hyperplasia (CAH). In this study, we generated a humanized 21-hydroxylase mouse model as the first step to the generation of mutant mice with different CAH-causing mutations. We replaced the mouse Cyp21a1 gene with the human CYP21A2 gene using homologous recombination in combination with CRISPR/Cas9 technique. The aim of this study was to characterize the new humanized mouse model. All results described are related to the homozygous animals in comparison with wild-type mice. We show analogous expression patterns of human 21-hydroxylase by the murine promoter and regulatory elements in comparison to murine 21-hydroxylase in wild-type animals. As expected, no Cyp21a1 transcript was detected in homozygous CYP21A2 adrenal glands. Alterations in adrenal gene expression were observed for Cyp11a1, Star, and Cyb11b1. These differences, however, were not pathological. Outward appearance, viability, growth, and fertility were not affected in the humanized CYP21A2 mice. Plasma steroid levels of corticosterone and aldosterone showed no pathological reduction. In addition, adrenal gland morphology and zonation were similar in both the humanized and the wild-type mice. In conclusion, humanized homozygous CYP21A2 mice developed normally and showed no differences in histological analyses, no reduction in adrenal and gonadal gene expression, or in plasma steroids in comparison with wild-type littermates.
2021
Lei Xing, Agnieszka Kubik-Zahorodna, Takashi Namba, Anneline Pinson, Marta Florio, Jan Prochazka, Mihail Sarov, Radislav Sedlacek, Wieland Huttner Expression of human-specific ARHGAP11B in mice leads to neocortex expansion and increased memory flexibility. EMBO J, 40(13) Art. No. 107093 (2021)
Open Access DOI
Neocortex expansion during human evolution provides a basis for our enhanced cognitive abilities. Yet, which genes implicated in neocortex expansion are actually responsible for higher cognitive abilities is unknown. The expression of human-specific ARHGAP11B in embryonic/foetal mouse, ferret and marmoset neocortex was previously found to promote basal progenitor proliferation, upper-layer neuron generation and neocortex expansion during development, features commonly thought to contribute to increased cognitive abilities. However, a key question is whether this phenotype persists into adulthood and if so, whether cognitive abilities are indeed increased. Here, we generated a transgenic mouse line with physiological ARHGAP11B expression that exhibits increased neocortical size and upper-layer neuron numbers persisting into adulthood. Adult ARHGAP11B-transgenic mice showed altered neurobehaviour, notably increased memory flexibility and a reduced anxiety level. Our data are consistent with the notion that neocortex expansion by ARHGAP11B, a gene implicated in human evolution, underlies some of the altered neurobehavioural features observed in the transgenic mice, such as the increased memory flexibility, a neocortex-associated trait, with implications for the increase in cognitive abilities during human evolution.
Andreas Lackner*, Robert Sehlke*, Marius Garmhausen*, Giuliano Giuseppe Stirparo*, Michelle Huth, Fabian Titz-Teixeira, Petra van der Lelij, Julia Ramesmayer, Henry F Thomas, Meryem Ralser, Laura Santini, Elena Galimberti, Mihail Sarov, A F Stewart, Austin Smith, Andreas Beyer, Martin Leeb Cooperative genetic networks drive embryonic stem cell transition from naïve to formative pluripotency. EMBO J, 40(8) Art. No. e105776 (2021)
Open Access DOI
In the mammalian embryo, epiblast cells must exit the naïve state and acquire formative pluripotency. This cell state transition is recapitulated by mouse embryonic stem cells (ESCs), which undergo pluripotency progression in defined conditions in vitro. However, our understanding of the molecular cascades and gene networks involved in the exit from naïve pluripotency remains fragmentary. Here, we employed a combination of genetic screens in haploid ESCs, CRISPR/Cas9 gene disruption, large-scale transcriptomics and computational systems biology to delineate the regulatory circuits governing naïve state exit. Transcriptome profiles for 73 ESC lines deficient for regulators of the exit from naïve pluripotency predominantly manifest delays on the trajectory from naïve to formative epiblast. We find that gene networks operative in ESCs are also active during transition from pre- to post-implantation epiblast in utero. We identified 496 naïve state-associated genes tightly connected to the in vivo epiblast state transition and largely conserved in primate embryos. Integrated analysis of mutant transcriptomes revealed funnelling of multiple gene activities into discrete regulatory modules. Finally, we delineate how intersections with signalling pathways direct this pivotal mammalian cell state transition.
2020
Juliana G. Roscito, Kaushikaram Subramanian, Ronald Naumann, Mihail Sarov, Anna Shevchenko, Aliona Bogdanova, Thomas Kurth, Leo Foerster, Moritz Kreysing, Michael Hiller Recapitulating evolutionary divergence in a single cis-regulatory element is sufficient to cause expression changes of the lens gene Tdrd7. Mol Biol Evol, 38(2) 380-392 (2020)
Open AccessPDF
DOI
Mutations in cis-regulatory elements play important roles for phenotypic changes during evolution. Eye degeneration in the blind mole rat (BMR; Nannospalax galili) and other subterranean mammals is significantly associated with widespread divergence of eye regulatory elements, but the effect of these regulatory mutations on eye development and function has not been explored. Here, we investigate the effect of mutations observed in the BMR sequence of a conserved non-coding element upstream of Tdrd7, a pleiotropic gene required for lens development and spermatogenesis. We first show that this conserved element is a transcriptional repressor in lens cells and that the BMR sequence partially lost repressor activity. Next, we recapitulated evolutionary changes in this element by precisely replacing the endogenous regulatory element in a mouse line by the orthologous BMR sequence with CRISPR-Cas9. Strikingly, this repressor replacement caused a more than two-fold up-regulation of Tdrd7 in the developing lens; however, increased mRNA level does not result in a corresponding increase in TDRD7 protein nor an obvious lens phenotype, possibly explained by buffering at the posttranscriptional level. Our results are consistent with eye degeneration in subterranean mammals having a polygenic basis where many small-effect mutations in different eye-regulatory elements collectively contribute to phenotypic differences.
Robert W Fernandez, Kimberly Wei, Erin Y Wang, Deimante Mikalauskaite, Andrew Olson, Judy Pepper, Nakeirah Christie, Seongseop Kim, Susanne Weissenborn, Mihail Sarov, Michael R Koelle Cellular Expression and Functional Roles of All 26 Neurotransmitter GPCRs in the C. elegans Egg-Laying Circuit. J Neurosci, 40(39) 7475-7488 (2020)
DOI
Maps of the synapses made and neurotransmitters released by all neurons in model systems such as C. elegans have left still unresolved how neural circuits integrate and respond to neurotransmitter signals. Using the egg-laying circuit of C. elegans as a model, we mapped which cells express each of the 26 neurotransmitter G protein coupled receptors (GPCRs) of this organism and also genetically analyzed the functions of all 26 GPCRs. We found that individual neurons express many distinct receptors, epithelial cells often express neurotransmitter receptors, and receptors are often positioned to receive extrasynaptic signals. Receptor knockouts reveal few egg-laying defects under standard lab conditions, suggesting the receptors function redundantly or regulate egg-laying only in specific conditions; however, increasing receptor signaling through overexpression more efficiently reveals receptor functions. This map of neurotransmitter GPCR expression and function in the egg-laying circuit provides a model for understanding GPCR signaling in other neural circuits.SIGNIFICANCE STATEMENTNeurotransmitters signal through G protein coupled receptors (GPCRs) to modulate activity of neurons, and changes in such signaling can underlie conditions such as depression and Parkinson's disease. To determine how neurotransmitter GPCRs together help regulate function of a neural circuit, we analyzed the simple egg-laying circuit in the model organism C. elegans. We identified all the cells that express every neurotransmitter GPCR and genetically analyzed how each GPCR affects the behavior the circuit produces. We found that many neurotransmitter GPCRs are expressed in each neuron, that neurons also appear to use these receptors to communicate with other cell types, and that GPCRs appear to often act redundantly or only under specific conditions to regulate circuit function.
Alexandra Lewis, Ahmet C Berkyurek, Andre Greiner, Ahilya N Sawh, Ajay A Vashisht, Stephanie Merrett, Mathieu N Flamand, James Wohlschlegel, Mihail Sarov, Eric A Miska, Thomas F Duchaine A Family of Argonaute-Interacting Proteins Gates Nuclear RNAi. Mol Cell, 78(5) 862-875 (2020)
DOI
Nuclear RNA interference (RNAi) pathways work together with histone modifications to regulate gene expression and enact an adaptive response to transposable RNA elements. In the germline, nuclear RNAi can lead to trans-generational epigenetic inheritance (TEI) of gene silencing. We identified and characterized a family of nuclear Argonaute-interacting proteins (ENRIs) that control the strength and target specificity of nuclear RNAi in C. elegans, ensuring faithful inheritance of epigenetic memories. ENRI-1/2 prevent misloading of the nuclear Argonaute NRDE-3 with small RNAs that normally effect maternal piRNAs, which prevents precocious nuclear translocation of NRDE-3 in the early embryo. Additionally, they are negative regulators of nuclear RNAi triggered from exogenous sources. Loss of ENRI-3, an unstable protein expressed mostly in the male germline, misdirects the RNAi response to transposable elements and impairs TEI. The ENRIs determine the potency and specificity of nuclear RNAi responses by gating small RNAs into specific nuclear Argonautes.
Sören Reinke, Mary Linge, Hans H Diebner, H Luksch, Silke Glage, Anne Gocht, Avril A B Robertson, Matthew A Cooper, Sigrun R Hofmann, Ronald Naumann, Mihail Sarov, Rayk Behrendt, Axel Roers, Frank Pessler, Joachim Roesler, Angela Rösen-Wolff, Stefan Winkler Non-canonical Caspase-1 Signaling Drives RIP2-Dependent and TNF-α-Mediated Inflammation In Vivo. Cell Rep, 30(8) 2501-2511 (2020)
Open Access DOI
Pro-inflammatory caspase-1 is a key player in innate immunity. Caspase-1 processes interleukin (IL)-1β and IL-18 to their mature forms and triggers pyroptosis. These caspase-1 functions are linked to its enzymatic activity. However, loss-of-function missense mutations in CASP1 do not prevent autoinflammation in patients, despite decreased IL-1β production. In vitro data suggest that enzymatically inactive caspase-1 drives inflammation via enhanced nuclear factor κB (NF-κB) activation, independent of IL-1β processing. Here, we report two mouse models of enzymatically inactive caspase-1-C284A, demonstrating the relevance of this pathway in vivo. In contrast to Casp1-/- mice, caspase-1-C284A mice show pronounced hypothermia and increased levels of the pro-inflammatory cytokines tumor necrosis factor alpha (TNF-α) and IL-6 when challenged with lipopolysaccharide (LPS). Caspase-1-C284A signaling is RIP2 dependent and mediated by TNF-α but independent of the NLRP3 inflammasome. LPS-stimulated whole blood from patients carrying loss-of-function missense mutations in CASP1 secretes higher amounts of TNF-α. Taken together, these results reveal non-canonical caspase-1 signaling in vivo.
2019
Aleksandra Spiegel, Mandy Bachmann, Gabriel Jurado Jiménez, Mihail Sarov CRISPR/Cas9-based knockout pipeline for reverse genetics in mammalian cell culture. Methods, 164/165 49-58 (2019)
Open Access DOI
We present a straightforward protocol for reverse genetics in cultured mammalian cells, using CRISPR/Cas9-mediated homology-dependent repair (HDR) based insertion of a protein trap cassette, resulting in a termination of the endogenous gene expression. Complete loss of function can be achieved with monoallelic trap cassette insertion, as the second allele is frequently disrupted by an error-prone non-homologous end joining (NHEJ) mechanism. The method should be applicable to any expressed gene in most cell lines, including those with low HDR efficiency, as the knockout alleles can be directly selected for.
2018
Samir Vaid, J Gray Camp, Lena Hersemann, Christina Eugster Oegema, Anne-Kristin Heninger, Sylke Winkler, Holger Brandl, Mihail Sarov, Barbara Treutlein, Wieland Huttner#, Takashi Namba# A novel population of Hopx-dependent basal radial glial cells in the developing mouse neocortex. Development, 145(20) Art. No. dev169276 (2018)
DOI
A specific subpopulation of neural progenitor cells, the basal radial glial cells (bRGCs) of the outer subventricular zone (OSVZ), are thought to have a key role in the evolutionary expansion of the mammalian neocortex. In the developing lissencephalic mouse neocortex, bRGCs exist at low abundance and show significant molecular differences from bRGCs in developing gyrencephalic species. Here, we demonstrate that the developing mouse medial neocortex (medNcx), in contrast to the canonically studied lateral neocortex (latNcx), exhibits an OSVZ and an abundance of bRGCs similar to that in developing gyrencephalic neocortex. Unlike bRGCs in developing mouse latNcx, the bRGCs in medNcx exhibit human bRGC-like gene expression, including expression of Hopx, a human bRGC marker. Disruption of Hopx expression in mouse embryonic medNcx and forced Hopx expression in mouse embryonic latNcx demonstrate that Hopx is required and sufficient, respectively, for bRGC abundance as found in the developing gyrencephalic neocortex. Taken together, our data identify a novel bRGC subpopulation in developing mouse medNcx that is highly related to bRGCs of developing gyrencephalic neocortex.
Yusuke Toyoda#, Busra Akarlar, Mihail Sarov, Nurhan Ozlu, Shigeaki Saitoh# Extracellular glucose level regulates dependence on GRP78 for cell surface localization of multipass transmembrane proteins in HeLa cells. FEBS Lett, 592(19) 3295-3304 (2018)
DOI
Many human-cultured cell lines survive glucose starvation, but the underlying mechanisms remain unclear. Here, we searched for proteins required for cellular adaptation to glucose-limited conditions and identified several endoplasmic reticulum chaperones in the glucose-regulated protein (GRP) family as proteins enriched in the cellular membrane. Surprisingly, these proteins, which are required for cell surface localization of GLUT1 under high-glucose conditions, become dispensable for targeting GLUT1 to the surface upon glucose starvation. In marked contrast, cell surface localization of single-pass transmembrane proteins, such as epidermal growth factor receptor and CD98, is not disturbed by GRP78 depletion regardless of the extracellular glucose level. These results indicate that the extracellular glucose level regulates dependence on the GRPs for cell surface localization of multipass transmembrane proteins.
Jan-Philip Medelnik, Kathleen Roensch, Satoshi Okawa, Antonio Del Sol, Osvaldo Chara, Levan Mchedlishvili, Elly M. Tanaka Signaling-Dependent Control of Apical Membrane Size and Self-Renewal in Rosette-Stage Human Neuroepithelial Stem Cells. Stem Cell Rep, 10(6) 1751-1765 (2018)
Open Access DOI
In the developing nervous system, neural stem cells are polarized and maintain an apical domain facing a central lumen. The presence of apical membrane is thought to have a profound influence on maintaining the stem cell state. With the onset of neurogenesis, cells lose their polarization, and the concomitant loss of the apical domain coincides with a loss of the stem cell identity. Little is known about the molecular signals controlling apical membrane size. Here, we use two neuroepithelial cell systems, one derived from regenerating axolotl spinal cord and the other from human embryonic stem cells, to identify a molecular signaling pathway initiated by lysophosphatidic acid that controls apical membrane size and consequently controls and maintains epithelial organization and lumen size in neuroepithelial rosettes. This apical domain size increase occurs independently of effects on proliferation and involves a serum response factor-dependent transcriptional induction of junctional and apical membrane components.
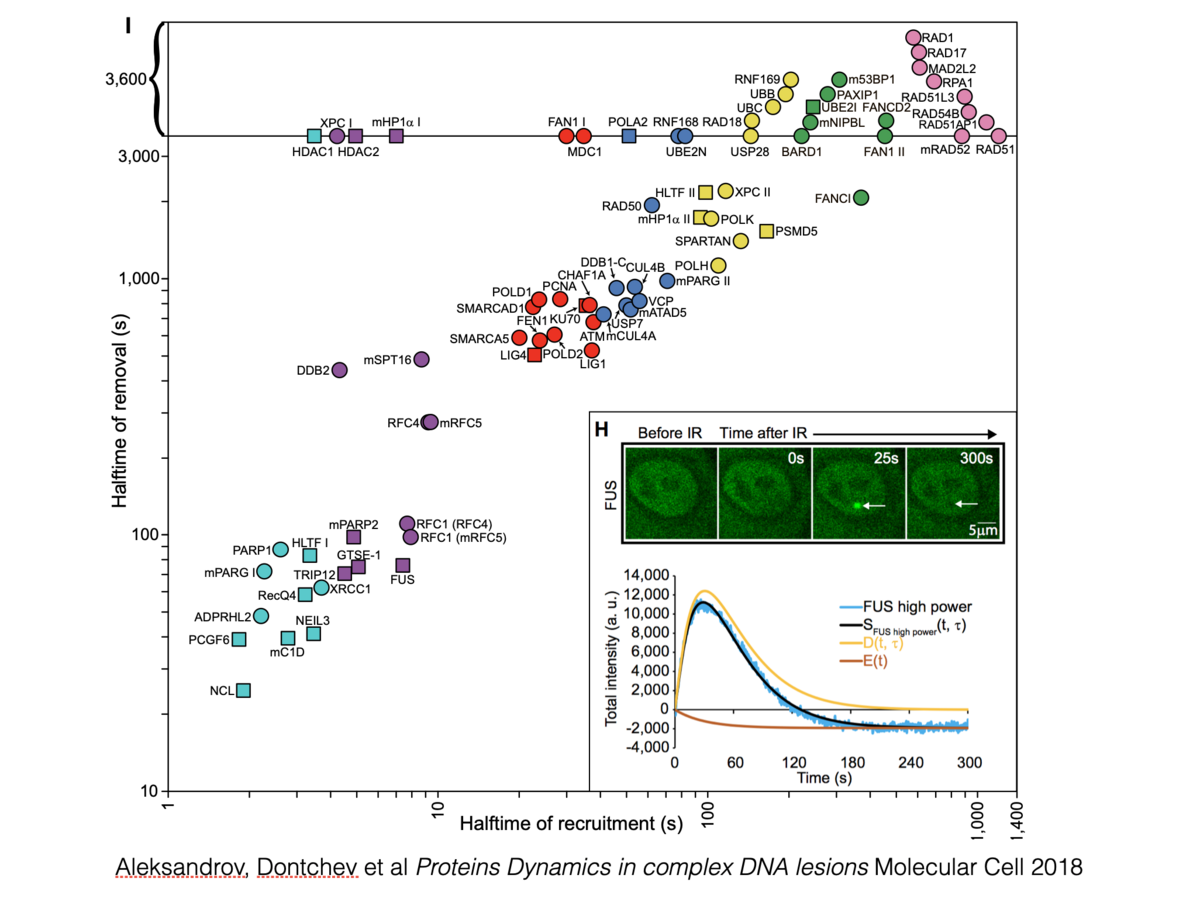
Radoslav Aleksandrov, Anton Dotchev, Ina Poser, Dragomir Krastev, Georgi Georgiev, Greta C Panova, Yordan Babukov, Georgi Danovski, Teodora Dyankova, Lars Hubatsch, Aneliya Ivanova, Aleksandar Atemin, Marina N Nedelcheva-Veleva, Susanne Hasse, Mihail Sarov, Frank Buchholz, Anthony Hyman, Stephan W. Grill, Stoyno Stoynov Protein Dynamics in Complex DNA Lesions. Mol Cell, 69(6) 1046-1061 (2018)
DOI
A single mutagen can generate multiple different types of DNA lesions. How different repair pathways cooperate in complex DNA lesions, however, remains largely unclear. Here we measured, clustered, and modeled the kinetics of recruitment and dissociation of 70 DNA repair proteins to laser-induced DNA damage sites in HeLa cells. The precise timescale of protein recruitment reveals that error-prone translesion polymerases are considerably delayed compared to error-free polymerases. We show that this is ensured by the delayed recruitment of RAD18 to double-strand break sites. The time benefit of error-free polymerases disappears when PARP inhibition significantly delays PCNA recruitment. Moreover, removal of PCNA from complex DNA damage sites correlates with RPA loading during 5'-DNA end resection. Our systematic study of the dynamics of DNA repair proteins in complex DNA lesions reveals the multifaceted coordination between the repair pathways and provides a kinetics-based resource to study genomic instability and anticancer drug impact.
2017
Edlyn Wu, Ajay A Vashisht, Clément Chapat, Mathieu N Flamand, Emiliano Cohen, Mihail Sarov, Yuval Tabach, Nahum Sonenberg, James Wohlschlegel, Thomas F Duchaine A continuum of mRNP complexes in embryonic microRNA-mediated silencing. Nucleic Acids Res, 45(4) 2081-2098 (2017)
Open AccessPDF
DOI
2016
Susanne Hasse, Anthony Hyman, Mihail Sarov TransgeneOmics - A transgenic platform for protein localization based function exploration. Methods, 96 69-74 (2016)
Open AccessPDF
DOI
The localization of a protein is intrinsically linked to its role in the structural and functional organization of the cell. Advances in transgenic technology have streamlined the use of protein localization as a function discovery tool. Here we review the use of large genomic DNA constructs such as bacterial artificial chromosomes as a transgenic platform for systematic tag-based protein function exploration.
Sadhna Phanse, Cuihong Wan, Blake Borgeson, Fan Tu, Kevin Drew, Greg Clark, Xuejian Xiong, Olga Kagan, Julian Kwan, Alexandr Bezginov, Kyle Chessman, Swati Pal, Graham Cromar, Ophelia Papoulas, Zuyao Ni, Daniel R Boutz, Snejana Stoilova, Pierre C Havugimana, Xinghua Guo, Ramy H Malty, Mihail Sarov, Jack Greenblatt, Mohan Babu, W Brent Derry, Elisabeth R Tillier, John Wallingford, John Parkinson, Edward M Marcotte, Andrew Emili Proteome-wide dataset supporting the study of ancient metazoan macromolecular complexes. Data Brief, 6 715-721 (2016)
Open AccessPDF
DOI
Our analysis examines the conservation of multiprotein complexes among metazoa through use of high resolution biochemical fractionation and precision mass spectrometry applied to soluble cell extracts from 5 representative model organisms Caenorhabditis elegans, Drosophila melanogaster, Mus musculus, Strongylocentrotus purpuratus, and Homo sapiens. The interaction network obtained from the data was validated globally in 4 distant species (Xenopus laevis, Nematostella vectensis, Dictyostelium discoideum, Saccharomyces cerevisiae) and locally by targeted affinity-purification experiments. Here we provide details of our massive set of supporting biochemical fractionation data available via ProteomeXchange (PXD002319-PXD002328), PPIs via BioGRID (185267); and interaction network projections via (http://metazoa.med.utoronto.ca) made fully accessible to allow further exploration. The datasets here are related to the research article on metazoan macromolecular complexes in Nature [1].
Nereo Kalebic, Elena Taverna, Stefania Tavano, Fong Kuan Wong, Dana Suchold, Sylke Winkler, Wieland B. Huttner, Mihail Sarov CRISPR/Cas9-induced disruption of gene expression in mouse embryonic brain and single neural stem cells in vivo. EMBO Rep, 17(3) 338-348 (2016)
Open AccessPDF
DOI
We have applied the CRISPR/Cas9 system in vivo to disrupt gene expression in neural stem cells in the developing mammalian brain. Two days after in utero electroporation of a single plasmid encoding Cas9 and an appropriate guide RNA (gRNA) into the embryonic neocortex of Tis21::GFP knock-in mice, expression of GFP, which occurs specifically in neural stem cells committed to neurogenesis, was found to be nearly completely (≈90%) abolished in the progeny of the targeted cells. Importantly, upon in utero electroporation directly of recombinant Cas9/gRNA complex, near-maximal efficiency of disruption of GFP expression was achieved already after 24 h. Furthermore, by using microinjection of the Cas9 protein/gRNA complex into neural stem cells in organotypic slice culture, we obtained disruption of GFP expression within a single cell cycle. Finally, we used either Cas9 plasmid in utero electroporation or Cas9 protein complex microinjection to disrupt the expression of Eomes/Tbr2, a gene fundamental for neocortical neurogenesis. This resulted in a reduction in basal progenitors and an increase in neuronal differentiation. Thus, the present in vivo application of the CRISPR/Cas9 system in neural stem cells provides a rapid, efficient and enduring disruption of expression of specific genes to dissect their role in mammalian brain development.
Mihail Sarov#, Christiane Barz, Helena Jambor, Marco Y Hein, Christopher Schmied, Dana Suchold, Bettina Stender, Stephan Janosch, Vinay Vikas Kj, R T Krishnan, Aishwarya Krishnamoorthy, Irene R S Ferreira, Radoslaw K Ejsmont, Katja Finkl, Susanne Hasse, Philipp Kämpfer, Nicole Plewka, Elisabeth Vinis, Siegfried Schloissnig, Elisabeth Knust, Volker Hartenstein, Matthias Mann, Mani Ramaswami, K VijayRaghavan, Pavel Tomancak#, Frank Schnorrer# A genome-wide resource for the analysis of protein localisation in Drosophila. Elife, 5 Art. No. e12068 (2016)
Open AccessPDF
DOI
The Drosophila genome contains >13,000 protein coding genes, the majority of which remain poorly investigated. Important reasons include the lack of antibodies or reporter constructs to visualise these proteins. Here we present a genome-wide fosmid library of 10,000 GFP-tagged clones, comprising tagged genes and most of their regulatory information. For 880 tagged proteins we created transgenic lines and for a total of 207 lines we assessed protein expression and localisation in ovaries, embryos, pupae or adults by stainings and live imaging approaches. Importantly, we visualised many proteins at endogenous expression levels and found a large fraction of them localising to subcellular compartments. By applying genetic complementation tests we estimate that about two-thirds of the tagged proteins are functional. Moreover, these tagged proteins enable interaction proteomics from developing pupae and adult flies. Taken together, this resource will boost systematic analysis of protein expression and localisation in various cellular and developmental contexts.
2015
Cuihong Wan, Blake Borgeson, Sadhna Phanse, Fan Tu, Kevin Drew, Greg Clark, Xuejian Xiong, Olga Kagan, Julian Kwan, Alexandr Bezginov, Kyle Chessman, Swati Pal, Graham Cromar, Ophelia Papoulas, Zuyao Ni, Daniel R Boutz, Snejana Stoilova, Pierre C Havugimana, Xinghua Guo, Ramy H Malty, Mihail Sarov, Jack Greenblatt, Mohan Babu, W Brent Derry, Elisabeth R Tillier, John Wallingford, John Parkinson, Edward M Marcotte, Andrew Emili Panorama of ancient metazoan macromolecular complexes. Nature, 525(7569) 339-344 (2015)
DOI
Macromolecular complexes are essential to conserved biological processes, but their prevalence across animals is unclear. By combining extensive biochemical fractionation with quantitative mass spectrometry, here we directly examined the composition of soluble multiprotein complexes among diverse metazoan models. Using an integrative approach, we generated a draft conservation map consisting of more than one million putative high-confidence co-complex interactions for species with fully sequenced genomes that encompasses functional modules present broadly across all extant animals. Clustering reveals a spectrum of conservation, ranging from ancient eukaryotic assemblies that have probably served cellular housekeeping roles for at least one billion years, ancestral complexes that have accrued contemporary components, and rarer metazoan innovations linked to multicellularity. We validated these projections by independent co-fractionation experiments in evolutionarily distant species, affinity purification and functional analyses. The comprehensiveness, centrality and modularity of these reconstructed interactomes reflect their fundamental mechanistic importance and adaptive value to animal cell systems.
Eric Cornes, Montserrat Porta-De-La-Riva, David Aristizábal-Corrales, Ana María Brokate-Llanos, Francisco Javier García-Rodríguez, Iris Ertl, Mònica Díaz, Laura Fontrodona, Kadri Reis, Robert Johnsen, David Baillie, Manuel J Muñoz, Mihail Sarov, Denis Dupuy, Julián Cerón Cytoplasmic LSM-1 protein regulates stress responses through the insulin/IGF-1 signaling pathway in Caenorhabditis elegans. RNA, 21(9) 1544-1553 (2015)
DOI
Genes coding for members of the Sm-like (LSm) protein family are conserved through evolution from prokaryotes to humans. These proteins have been described as forming homo- or heterocomplexes implicated in a broad range of RNA-related functions. To date, the nuclear LSm2-8 and the cytoplasmic LSm1-7 heteroheptamers are the best characterized complexes in eukaryotes. Through a comprehensive functional study of the LSm family members, we found that lsm-1 and lsm-3 are not essential for C. elegans viability, but their perturbation, by RNAi or mutations, produces defects in development, reproduction, and motility. We further investigated the function of lsm-1, which encodes the distinctive protein of the cytoplasmic complex. RNA-seq analysis of lsm-1 mutants suggests that they have impaired Insulin/IGF-1 signaling (IIS), which is conserved in metazoans and involved in the response to various types of stress through the action of the FOXO transcription factor DAF-16. Further analysis using a DAF-16::GFP reporter indicated that heat stress-induced translocation of DAF-16 to the nuclei is dependent on lsm-1. Consistent with this, we observed that lsm-1 mutants display heightened sensitivity to thermal stress and starvation, while overexpression of lsm-1 has the opposite effect. We also observed that under stress, cytoplasmic LSm proteins aggregate into granules in an LSM-1-dependent manner. Moreover, we found that lsm-1 and lsm-3 are required for other processes regulated by the IIS pathway, such as aging and pathogen resistance.
Maria L Spletter, Christiane Barz, Assa Yeroslaviz, Cornelia Schönbauer, Irene R S Ferreira, Mihail Sarov, Daniel Gerlach, Alexander Stark, Bianca Habermann, Frank Schnorrer The RNA-binding protein Arrest (Bruno) regulates alternative splicing to enable myofibril maturation in Drosophila flight muscle. EMBO Rep, 16(2) 178-191 (2015)
DOI
In Drosophila, fibrillar flight muscles (IFMs) enable flight, while tubular muscles mediate other body movements. Here, we use RNA-sequencing and isoform-specific reporters to show that spalt major (salm) determines fibrillar muscle physiology by regulating transcription and alternative splicing of a large set of sarcomeric proteins. We identify the RNA-binding protein Arrest (Aret, Bruno) as downstream of salm. Aret shuttles between the cytoplasm and nuclei and is essential for myofibril maturation and sarcomere growth of IFMs. Molecularly, Aret regulates IFM-specific splicing of various salm-dependent sarcomeric targets, including Stretchin and wupA (TnI), and thus maintains muscle fiber integrity. As Aret and its sarcomeric targets are evolutionarily conserved, similar principles may regulate mammalian muscle morphogenesis.
Sider Penkov, Damla Kaptan, Cihan Erkut, Mihail Sarov, Fanny Mende, Teymuras V. Kurzchalia Integration of carbohydrate metabolism and redox state controls dauer larva formation in Caenorhabditis elegans. Nat Commun, 6 Art. No. 8060 (2015)
DOI
Under adverse conditions, Caenorhabditis elegans enters a diapause stage called the dauer larva. External cues signal the nuclear hormone receptor DAF-12, the activity of which is regulated by its ligands: dafachronic acids (DAs). DAs are synthesized from cholesterol, with the last synthesis step requiring NADPH, and their absence stimulates dauer formation. Here we show that NADPH levels determine dauer formation in a regulatory mechanism involving key carbohydrate and redox metabolic enzymes. Elevated trehalose biosynthesis diverts glucose-6-phosphate from the pentose phosphate pathway, which is the major source of cellular NADPH. This enhances dauer formation due to the decrease in the DA level. Moreover, DAF-12, in cooperation with DAF-16/FoxO, induces negative feedback of DA synthesis via activation of the trehalose-producing enzymes TPS-1/2 and inhibition of the NADPH-producing enzyme IDH-1. Thus, the dauer developmental decision is controlled by integration of the metabolic flux of carbohydrates and cellular redox potential.
2014
Christian Frøkjær-Jensen, M Wayne Davis, Mihail Sarov, Jon Taylor, Stephane Flibotte, Matthew LaBella, Andrei I. Pozniakovsky, Donald G Moerman, Erik M Jorgensen Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nat Methods, 11(5) 529-534 (2014)
DOI
We have generated a recombinant Mos1 transposon that can insert up to 45-kb transgenes into the Caenorhabditis elegans genome. The minimal Mos1 transposon (miniMos) is 550 bp long and inserts DNA into the genome at high frequency (~60% of injected animals). Genetic and antibiotic markers can be used for selection, and the transposon is active in C. elegans isolates and Caenorhabditis briggsae. We used the miniMos transposon to generate six universal Mos1-mediated single-copy insertion (mosSCI) landing sites that allow targeted transgene insertion with a single targeting vector into permissive expression sites on all autosomes. We also generated two collections of strains: a set of bright fluorescent insertions that are useful as dominant, genetic balancers and a set of lacO insertions to track genome position.
Stephan Preibisch#, Fernando Amat, Evangelia Stamataki, Mihail Sarov, Robert H. Singer, Gene Myers, Pavel Tomancak# Efficient Bayesian-based multiview deconvolution. Nat Methods, 11(6) 645-648 (2014)
PDF
DOI
Light-sheet fluorescence microscopy is able to image large specimens with high resolution by capturing the samples from multiple angles. Multiview deconvolution can substantially improve the resolution and contrast of the images, but its application has been limited owing to the large size of the data sets. Here we present a Bayesian-based derivation of multiview deconvolution that drastically improves the convergence time, and we provide a fast implementation using graphics hardware.
Indulekha P Sudhakaran, Jens Hillebrand, Adrian Dervan, Shradha Das, Eimear E Holohan, Jörn Hülsmeier, Mihail Sarov, Roy Parker, K VijayRaghavan, Mani Ramaswami FMRP and Ataxin-2 function together in long-term olfactory habituation and neuronal translational control. Proc Natl Acad Sci U.S.A., 111(1) 99-108 (2014)
DOI
Fragile X mental retardation protein (FMRP) and Ataxin-2 (Atx2) are triplet expansion disease- and stress granule-associated proteins implicated in neuronal translational control and microRNA function. We show that Drosophila FMRP (dFMR1) is required for long-term olfactory habituation (LTH), a phenomenon dependent on Atx2-dependent potentiation of inhibitory transmission from local interneurons (LNs) to projection neurons (PNs) in the antennal lobe. dFMR1 is also required for LTH-associated depression of odor-evoked calcium transients in PNs. Strong transdominant genetic interactions among dFMR1, atx2, the deadbox helicase me31B, and argonaute1 (ago1) mutants, as well as coimmunoprecitation of dFMR1 with Atx2, indicate that dFMR1 and Atx2 function together in a microRNA-dependent process necessary for LTH. Consistently, PN or LN knockdown of dFMR1, Atx2, Me31B, or the miRNA-pathway protein GW182 increases expression of a Ca2+/calmodulin-dependent protein kinase II (CaMKII) translational reporter. Moreover, brain immunoprecipitates of dFMR1 and Atx2 proteins include CaMKII mRNA, indicating respective physical interactions with this mRNA. Because CaMKII is necessary for LTH, these data indicate that fragile X mental retardation protein and Atx2 act via at least one common target RNA for memory-associated long-term synaptic plasticity. The observed requirement in LNs and PNs supports an emerging view that both presynaptic and postsynaptic translation are necessary for long-term synaptic plasticity. However, whereas Atx2 is necessary for the integrity of dendritic and somatic Me31B-containing particles, dFmr1 is not. Together, these data indicate that dFmr1 and Atx2 function in long-term but not short-term memory, regulating translation of at least some common presynaptic and postsynaptic target mRNAs in the same cells.
2013
Anna Ivanova, Yannis Kalaidzidis, Ronald Dirkx, Mihail Sarov, Michael Gerlach, Britta Schroth-Diez, Andreas Müller, Yanmei Liu, Cordula Andree, Bernard Mulligan, Carla Münster, Thomas Kurth, Marc Bickle, Stephan Speier, Konstantinos Anastassiadis, Michele Solimena Age-dependent labeling and imaging of insulin secretory granules. Diabetes, 62(11) 3687-3696 (2013)
DOI
Insulin is stored within the secretory granules of pancreatic β-cells, and impairment of its release is the hallmark of type 2 diabetes. Preferential exocytosis of newly synthesized insulin suggests that granule aging is a key factor influencing insulin secretion. Here, we illustrate a technology that enables the study of granule aging in insulinoma cells and β-cells of knock-in mice through the conditional and unequivocal labeling of insulin fused to the SNAP tag. This approach, which overcomes the limits encountered with previous strategies based on radiolabeling or fluorescence timer proteins, allowed us to formally demonstrate the preferential release of newly synthesized insulin and reveal that the motility of cortical granules significantly changes over time. Exploitation of this approach may enable the identification of molecular signatures associated with granule aging and unravel possible alterations of granule turnover in diabetic β-cells. Furthermore, the method is of general interest for the study of membrane traffic and aging.
Marina N Nedelcheva-Veleva, Mihail Sarov, Ivan Yanakiev, Eva Mihailovska, Miroslav P Ivanov, Greta C Panova, Stoyno Stoynov The thermodynamic patterns of eukaryotic genes suggest a mechanism for intron-exon recognition. Nat Commun, 4 Art. No. 2101 (2013)
DOI
The essential cis- and trans-acting elements required for RNA splicing have been defined, however, the detailed molecular mechanisms underlying intron-exon recognition are still unclear. Here we demonstrate that the ratio between stability of mRNA/DNA and DNA/DNA duplexes near 3'-spice sites is a characteristic feature that can contribute to intron-exon differentiation. Remarkably, throughout all transcripts, the most unstable mRNA/DNA duplexes, compared with the corresponding DNA/DNA duplexes, are situated upstream of the 3'-splice sites and include the polypyrimidine tracts. This characteristic instability is less pronounced in weak alternative splice sites and disease-associated cryptic 3'-splice sites. Our results suggest that this thermodynamic pattern can prevent the re-annealing of mRNA to the DNA template behind the RNA polymerase to ensure access of the splicing machinery to the polypyrimidine tract and the branch point. In support of this mechanism, we demonstrate that RNA/DNA duplex formation at this region prevents pre-spliceosome A complex assembly.
Claes Andréasson, Anna J Schick, Susanne M Pfeiffer, Mihail Sarov, Francis Stewart, Wolfgang Wurst, Joel A Schick Direct cloning of isogenic murine DNA in yeast and relevance of isogenicity for targeting in embryonic stem cells PLoS ONE, 8(9) Art. No. e74207 (2013)
Open Access DOI
Efficient gene targeting in embryonic stem cells requires that modifying DNA sequences are identical to those in the targeted chromosomal locus. Yet, there is a paucity of isogenic genomic clones for human cell lines and PCR amplification cannot be used in many mutation-sensitive applications. Here, we describe a novel method for the direct cloning of genomic DNA into a targeting vector, pRTVIR, using oligonucleotide-directed homologous recombination in yeast. We demonstrate the applicability of the method by constructing functional targeting vectors for mammalian genes Uhrf1 and Gfap. Whereas the isogenic targeting of the gene Uhrf1 showed a substantial increase in targeting efficiency compared to non-isogenic DNA in mouse E14 cells, E14-derived DNA performed better than the isogenic DNA in JM8 cells for both Uhrf1 and Gfap. Analysis of 70 C57BL/6-derived targeting vectors electroporated in JM8 and E14 cell lines in parallel showed a clear dependence on isogenicity for targeting, but for three genes isogenic DNA was found to be inhibitory. In summary, this study provides a straightforward methodological approach for the direct generation of isogenic gene targeting vectors.
2012
Mihail Sarov, John I Murray, Kristin Schanze, Andrei Pozniakovski, Wei Niu, Karolin Angermann, Susanne Hasse, Michaela Rupprecht, Elisabeth Vinis, Matthew Tinney, Elicia A. Preston, Andrea Zinke, Susanne Enst, Tina Teichgraber, Judith Janette, Kadri Reis, Stephan Janosch, Siegfried Schloissnig, Radoslaw K Ejsmont, Cindie Slightam, Xiao Xu, Stuart K Kim, Valerie Reinke, A Francis Stewart, Michael Snyder, Robert H Waterston, Anthony A. Hyman A Genome-Scale Resource for In Vivo Tag-Based Protein Function Exploration in C. elegans. Cell, 150(4) 855-866 (2012)
PDF
DOI
Understanding the in vivo dynamics of protein localization and their physical interactions is important for many problems in biology. To enable systematic protein function interrogation in a multicellular context, we built a genome-scale transgenic platform for in vivo expression of fluorescent- and affinity-tagged proteins in Caenorhabditis elegans under endogenous cis regulatory control. The platform combines computer-assisted transgene design, massively parallel DNA engineering, and next-generation sequencing to generate a resource of 14,637 genomic DNA transgenes, which covers 73% of the proteome. The multipurpose tag used allows any protein of interest to be localized in vivo or affinity purified using standard tag-based assays. We illustrate the utility of the resource by systematic chromatin immunopurification and automated 4D imaging, which produced detailed DNA binding and cell/tissue distribution maps for key transcription factor proteins.
2011
Marta Nedelkova, Marcello Maresca, Jun Fu, Mariya Rostovskaya, Ramu Chenna, Christian Thiede, Konstantinos Anastassiadis, Mihail Sarov, A Francis Stewart Targeted isolation of cloned genomic regions by recombineering for haplotype phasing and isogenic targeting. Nucleic Acids Res, 39(20) Art. No. e137 (2011)
DOI
Studying genetic variations in the human genome is important for understanding phenotypes and complex traits, including rare personal variations and their associations with disease. The interpretation of polymorphisms requires reliable methods to isolate natural genetic variations, including combinations of variations, in a format suitable for downstream analysis. Here, we describe a strategy for targeted isolation of large regions (∼35 kb) from human genomes that is also applicable to any genome of interest. The method relies on recombineering to fish out target fosmid clones from pools and thereby circumvents the laborious need to plate and screen thousands of individual clones. To optimize the method, a new highly recombineering-efficient bacterial host, including inducible TrfA for fosmid copy number amplification, was developed. Various regions were isolated from human embryonic stem cell lines and a personal genome, including highly repetitive and duplicated ones. The maternal and paternal alleles at the MECP2/IRAK 1 loci were distinguished based on identification of novel allele-specific single-nucleotide polymorphisms in regulatory regions. Additionally, we applied further recombineering to construct isogenic targeting vectors for patient-specific applications. These methods will facilitate work to understand the linkage between personal variations and disease propensity, as well as possibilities for personal genome surgery.
Antigoni Elefsinioti, Ömer Sinan Saraç, Anna Hegele, Conrad Plake, Nina C Hubner, Ina Poser, Mihail Sarov, Anthony A. Hyman, Matthias Mann, Michael Schroeder, Ulrich Stelzl, Andreas Beyer Large-scale de novo prediction of physical protein-protein association. Mol Cell Proteomics, 10(11) Art. No. M111.010629 (2011)
PDF
DOI
Information about the physical association of proteins is extensively used for studying cellular processes and disease mechanisms. However, complete experimental mapping of the human interactome will remain prohibitively difficult in the near future. Here we present a map of predicted human protein interactions that distinguishes functional association from physical binding. Our network classifies more than 5 million protein pairs predicting 94,009 new interactions with high confidence. We experimentally tested a subset of these predictions using yeast two-hybrid analysis and affinity purification followed by quantitative mass spectrometry. Thus we identified 462 new protein-protein interactions and confirmed the predictive power of the network. These independent experiments address potential issues of circular reasoning and are a distinctive feature of this work. Analysis of the physical interactome unravels subnetworks mediating between different functional and physical subunits of the cell. Finally, we demonstrate the utility of the network for the analysis of molecular mechanisms of complex diseases by applying it to genome-wide association studies of neurodegenerative diseases. This analysis provides new evidence implying TOMM40 as a factor involved in Alzheimer's disease. The network provides a high-quality resource for the analysis of genomic data sets and genetic association studies in particular. Our interactome is available via the hPRINT web server at: www.print-db.org.
Sarah C Petersen, Joseph D Watson, Janet E Richmond, Mihail Sarov, Walter W Walthall, David M Miller A transcriptional program promotes remodeling of GABAergic synapses in Caenorhabditis elegans J Neurosci, 31(43) 15362-15375 (2011)
DOI
Although transcription factors are known to regulate synaptic plasticity, downstream genes that contribute to neural circuit remodeling are largely undefined. In Caenorhabditis elegans, GABAergic Dorsal D (DD) motor neuron synapses are relocated to new sites during larval development. This remodeling program is blocked in Ventral D (VD) GABAergic motor neurons by the COUP-TF (chicken ovalbumin upstream promoter transcription factor) homolog, UNC-55. We exploited this UNC-55 function to identify downstream synaptic remodeling genes that encode a diverse array of protein types including ion channels, cytoskeletal components, and transcription factors. We show that one of these targets, the Iroquois-like homeodomain protein, IRX-1, functions as a key regulator of remodeling in DD neurons. Our discovery of irx-1 as an unc-55-regulated target defines a transcriptional pathway that orchestrates an intricate synaptic remodeling program. Moreover, the well established roles of these conserved transcription factors in mammalian neural development suggest that a similar cascade may also control synaptic plasticity in more complex nervous systems.
Helmut Hofemeister, Giovanni Ciotta, Jun Fu, Philipp Martin Seibert, Alexander Schulz, Marcello Maresca, Mihail Sarov, Konstantinos Anastassiadis, A. Francis Stewart Recombineering, transfection, Western, IP and ChIP methods for protein tagging via gene targeting or BAC transgenesis. Methods, 53(4) 437-452 (2011)
PDF
DOI
Protein tagging offers many advantages for proteomic and regulomic research. Ideally, protein tagging is equivalent to having a high affinity antibody for every chosen protein. However, these advantages are compromised if the tagged protein is overexpressed, which is usually the case from cDNA expression vectors. Physiological expression of tagged proteins can be achieved by gene targeting to knock-in the protein tag or by BAC transgenesis. BAC transgenes usually retain the native gene architecture including all cis-regulatory elements as well as the exon-intron configurations. Consequently most BAC transgenes are authentically regulated (e.g. by transcription factors, cell cycle, miRNA) and can be alternatively spliced. Recombineering has become the method of choice for generating targeting constructs or modifying BACs. Here we present methods with detailed protocols for protein tagging by recombineering for BAC transgenesis and/or gene targeting, including the evaluation of tagged protein expression, the retrieval of associated protein complexes for mass spectrometry and the use of the tags in ChIP (chromatin immunoprecipitation).
Giovanni Ciotta, Helmut Hofemeister, Marcello Maresca, Jun Fu, Mihail Sarov, Konstantinos Anastassiadis, A Francis Stewart Recombineering BAC transgenes for protein tagging Methods, 53(2) 113-119 (2011)
DOI
Protein tagging offers many advantages for proteomic and regulomic research, particularly due to the use of generic and highly sensitive methods that can be applied with reasonable throughput. Ideally, protein tagging is equivalent to having a high affinity antibody for every chosen protein. However, these advantages are compromised if the tagged protein is overexpressed, which is usually the case from cDNA expression vectors. BAC (bacterial artificial chromosome) transgenes present a way to express a chosen protein at physiological levels with all regulatory elements in their native configurations, including cell cycle, alternative splicing and microRNA regulation. Recombineering has become the method of choice for modifying large constructs like BACs. Here, we present a method for protein tagging by recombineering BACs, transfecting cells and evaluating tagged protein expression.
Wei Niu, Zhi John Lu, Mei Zhong, Mihail Sarov, James T Murray, Cathleen M Brdlik, Judith Janette, Chao Chen, Pedro Alves, Elicia A. Preston, Cindie Slightam, Lixia Jiang, Anthony A. Hyman, Stuart K Kim, Robert H Waterston, Mark Gerstein, Michael Snyder, Valerie Reinke Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res, 21(2) 245-254 (2011)
PDF
DOI
Regulation of gene expression by sequence-specific transcription factors is central to developmental programs and depends on the binding of transcription factors with target sites in the genome. To date, most such analyses in Caenorhabditis elegans have focused on the interactions between a single transcription factor with one or a few select target genes. As part of the modENCODE Consortium, we have used chromatin immunoprecipitation coupled with high-throughput DNA sequencing (ChIP-seq) to determine the genome-wide binding sites of 22 transcription factors (ALR-1, BLMP-1, CEH-14, CEH-30, EGL-27, EGL-5, ELT-3, EOR-1, GEI-11, HLH-1, LIN-11, LIN-13, LIN-15B, LIN-39, MAB-5, MDL-1, MEP-1, PES-1, PHA-4, PQM-1, SKN-1, and UNC-130) at diverse developmental stages. For each factor we determined candidate gene targets, both coding and non-coding. The typical binding sites of almost all factors are within a few hundred nucleotides of the transcript start site. Most factors target a mixture of coding and non-coding target genes, although one factor preferentially binds to non-coding RNA genes. We built a regulatory network among the 22 factors to determine their functional relationships to each other and found that some factors appear to act preferentially as regulators and others as target genes. Examination of the binding targets of three related HOX factors-LIN-39, MAB-5, and EGL-5-indicates that these factors regulate genes involved in cellular migration, neuronal function, and vulval differentiation, consistent with their known roles in these developmental processes. Ultimately, the comprehensive mapping of transcription factor binding sites will identify features of transcriptional networks that regulate C. elegans developmental processes.
Radoslaw K Ejsmont, Peter Ahlfeld, Andrei I. Pozniakovsky, A Francis Stewart, Pavel Tomancak#, Mihail Sarov# Recombination-mediated genetic engineering of large genomic DNA transgenes. Methods Mol Biol, 772 445-458 (2011)
PDF
DOI
Faithful gene activity reporters are a useful tool for evo-devo studies enabling selective introduction of specific loci between species and assaying the activity of large gene regulatory sequences. The use of large genomic constructs such as BACs and fosmids provides an efficient platform for exploration of gene function under endogenous regulatory control. Despite their large size they can be easily engineered using in vivo homologous recombination in Escherichia coli (recombineering). We have previously demonstrated that the efficiency and fidelity of recombineering are sufficient to allow high-throughput transgene engineering in liquid culture, and have successfully applied this approach in several model systems. Here, we present a detailed protocol for recombineering of BAC/fosmid transgenes for expression of fluorescent or affinity tagged proteins in Drosophila under endogenous in vivo regulatory control. The tag coding sequence is seamlessly recombineered into the genomic region contained in the BAC/fosmid clone, which is then integrated into the fly genome using ?C31 recombination. This protocol can be easily adapted to other recombineering projects.
2010
Mark Gerstein*, Zhi John Lu*, Eric L. Van Nostrand*, Chao Cheng*, Bradley I. Arshinoff*, Tao Liu*, Kevin Y. Yip*, Rebecca Robilotto*, Andreas Rechtsteiner*, Kohta Ikegami*, Pedro Alves*, Aurelien Chateigner*, Marc Perry*, Mitzi Morris*, Raymond Auerbach*, Xin Feng*, Jing Leng*, Anne Vielle*, Wei Niu*, Kahn Rhrissorrakrai*, Ashish Agarwal, Roger P. Alexander, Galt Barber, Cathleen M Brdlik, Jennifer Brennan, Jeremy Jean Brouillet, Adrian Carr, Ming-Sin Cheung, Hiram Clawson, Sergio Contrino, Luke O. Dannenberg, Abby F. Dernburg, Arshad Desai, Lindsay Dick, Andréa Dosé, Jiang Du, Thea Egelhofer, Sevinc Ercan, Ghia Euskirchen, Brent Ewing, Elise A. Feingold, Reto Gassmann, Peter J. Good, Phil Green, Francois Gullier, Michelle Gutwein, Mark S. Guyer, Lukas Habegger, Ting Han, Jorja G. Henikoff, Stefan R. Henz, Angie Hinrichs, Heather Holster, Anthony A. Hyman, A. Leo Iniguez, Judith Janette, Morten Jensen, Masaomi Kato, W. James Kent, Ellen Kephart, Vishal Khivansara, Ekta Khurana, John K. Kim, Paulina Kolasinska-Zwierz, Eric C. Lai, Isabel Latorre, Amber Leahey, Suzanna E Lewis, Paul Lloyd, Lucas Lochovsky, Rebecca F. Lowdon, Yaniv Lubling, Rachel Lyne, Michael MacCoss, Sebastian D. Mackowiak, Marco Mangone, Sheldon McKay, Desirea Mecenas, Gennifer Merrihew, David M. Miller, Andrew Muroyama, John I. Murray, Siew-Loon Ooi, Hoang Pham, Taryn Phippen, Elicia A. Preston, Nikolaus Rajewski, Gunnar Rätsch, Heidi Rosenbaum, Joel Rozowksy, Kim Rutherford, Peter Ruzanov, Mihail Sarov, Rajkumar Sasidharan, Andrea Sboner, Paul Scheid, Eran Segal, Hyunjin Shin, Chong Shou, Frank J. Slack, Cindie Slightam, Richard Smith, William C. Spencer, E. O. Stinson, Scott Taing, Teruaki Takasaki, Dionne Vafeados, Ksenia Voronina, Guilin Wang, Nicole L. Washington, Christina M. Whittle, Beijing Wu, Koon-Kiu Yan, Georg Zeller, Zheng Zha, Mei Zhong, Xingliang Zhou, Julie Ahringer, Susan Strome, Kristin C. Gunsalus, Gos Micklem, X. Shirley Liu, Valerie Reinke, Stuart K Kim, LaDeana W Hillier, Steven Henikoff, Fabio Piano, Michael Snyder, Lincoln Stein, Jason D. Lieb, Robert H Waterston Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project Science, 330(6012) 1775-1787 (2010)
PDF
We systematically generated large-scale data sets to improve genome annotation for the nematode Caenorhabditis elegans, a key model organism. These data sets include transcriptome profiling across a developmental time course, genome-wide identification of transcription factor–binding sites, and maps of chromatin organization. From this, we created more complete and accurate gene models, including alternative splice forms and candidate noncoding RNAs. We constructed hierarchical networks of transcription factor–binding and microRNA interactions and discovered chromosomal locations bound by an unusually large number of transcription factors. Different patterns of chromatin composition and histone modification were revealed between chromosome arms and centers, with similarly prominent differences between autosomes and the X chromosome. Integrating data types, we built statistical models relating chromatin, transcription factor binding, and gene expression. Overall, our analyses ascribed putative functions to most of the conserved genome.
Haiyan Lei, Tetsunari Fukushige, Wei Niu, Mihail Sarov, Valerie Reinke, Michael Krause A widespread distribution of genomic CeMyoD binding sites revealed and cross validated by ChIP-Chip and ChIP-Seq techniques. PLoS ONE, 5(12) Art. No. e15898 (2010)
PDF
DOI
Identifying transcription factor binding sites genome-wide using chromatin immunoprecipitation (ChIP)-based technology is becoming an increasingly important tool in addressing developmental questions. However, technical problems associated with factor abundance and suitable ChIP reagents are common obstacles to these studies in many biological systems. We have used two completely different, widely applicable methods to determine by ChIP the genome-wide binding sites of the master myogenic regulatory transcription factor HLH-1 (CeMyoD) in C. elegans embryos. The two approaches, ChIP-seq and ChIP-chip, yield strongly overlapping results revealing that HLH-1 preferentially binds to promoter regions of genes enriched for E-box sequences (CANNTG), known binding sites for this well-studied class of transcription factors. HLH-1 binding sites were enriched upstream of genes known to be expressed in muscle, consistent with its role as a direct transcriptional regulator. HLH-1 binding was also detected at numerous sites unassociated with muscle gene expression, as has been previously described for its mouse homolog MyoD. These binding sites may reflect several additional functions for HLH-1, including its interactions with one or more co-factors to activate (or repress) gene expression or a role in chromatin organization distinct from direct transcriptional regulation of target genes. Our results also provide a comparison of ChIP methodologies that can overcome limitations commonly encountered in these types of studies while highlighting the complications of assigning in vivo functions to identified target sites.
James R A Hutchins*, Yusuke Toyoda*, Björn Hegemann*, Ina Poser*, Jean-Karim Hériché, Martina M Sykora, Martina Augsburg, Otto Hudecz, Bettina A Buschhorn, Jutta Bulkescher, Christian Conrad, David Comartin, Alexander Schleiffer, Mihail Sarov, Andrei I. Pozniakovsky, Mikolaj Slabicki, Siegfried Schloissnig, Ines Steinmacher, Marit Leuschner, Andrea Ssykor, Steffen Lawo, Laurence Pelletier, Holger Stark, Kim Nasmyth, Jan Ellenberg, Richard Durbin, Frank Buchholz, Karl Mechtler, Anthony A. Hyman#, Jan-Michael Peters# Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science, 328(5978) 593-599 (2010)
PDF
DOI
Chromosome segregation and cell division are essential, highly ordered processes that depend on numerous protein complexes. Results from recent RNA interference screens indicate that the identity and composition of these protein complexes is incompletely understood. Using gene tagging on bacterial artificial chromosomes, protein localization, and tandem-affinity purification-mass spectrometry, the MitoCheck consortium has analyzed about 100 human protein complexes, many of which had not or had only incompletely been characterized. This work has led to the discovery of previously unknown, evolutionarily conserved subunits of the anaphase-promoting complex and the gamma-tubulin ring complex--large complexes that are essential for spindle assembly and chromosome segregation. The approaches we describe here are generally applicable to high-throughput follow-up analyses of phenotypic screens in mammalian cells.
Mei Zhong, Wei Niu, Zhi John Lu, Mihail Sarov, James T Murray, Judith Janette, Debasish Raha, Karyn L Sheaffer, Hugo Y K Lam, Elicia A. Preston, Cindie Slightam, LaDeana W Hillier, Trisha Brock, Ashish Agarwal, Raymond Auerbach, Anthony A. Hyman, Mark Gerstein, Susan E Mango, Stuart K Kim, Robert H Waterston, Valerie Reinke#, Michael Snyder# Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet, 6(2) Art. No. e1000848 (2010)
PDF
DOI
Transcription factors are key components of regulatory networks that control development, as well as the response to environmental stimuli. We have established an experimental pipeline in Caenorhabditis elegans that permits global identification of the binding sites for transcription factors using chromatin immunoprecipitation and deep sequencing. We describe and validate this strategy, and apply it to the transcription factor PHA-4, which plays critical roles in organ development and other cellular processes. We identified thousands of binding sites for PHA-4 during formation of the embryonic pharynx, and also found a role for this factor during the starvation response. Many binding sites were found to shift dramatically between embryos and starved larvae, from developmentally regulated genes to genes involved in metabolism. These results indicate distinct roles for this regulator in two different biological processes and demonstrate the versatility of transcription factors in mediating diverse biological roles.
2009
Radoslaw K Ejsmont, Mihail Sarov, Sylke Winkler, Kamil A Lipinski, Pavel Tomancák A toolkit for high-throughput, cross-species gene engineering in Drosophila. Nat Methods, 6(6) 435-437 (2009)
PDF
DOI
We generated two complementary genomic fosmid libraries for Drosophila melanogaster and Drosophila pseudoobscura that permit seamless modification of large genomic clones by high-throughput recombineering and direct transgenesis. The fosmid transgenes recapitulated endogenous gene expression patterns. These libraries, in combination with recombineering technology, will be useful to rescue mutant phenotypes, allow imaging of gene products in living flies and enable systematic analysis and manipulation of gene activity across species.
2008
Ina Poser, Mihail Sarov, James R A Hutchins, Jean-Karim Hériché, Yusuke Toyoda, Andrei I. Pozniakovsky, Daniela Weigl, Anja Nitzsche, Björn Hegemann, Alexander W. Bird, Laurence Pelletier, Ralf Kittler, Sujun Hua, Ronald Naumann, Martina Augsburg, Martina M Sykora, Helmut Hofemeister, Youming Zhang, Kim Nasmyth, Kevin P White, Steffen Dietzel, Karl Mechtler, Richard Durbin, A. Francis Stewart, Jan-Michael Peters, Frank Buchholz, Anthony A. Hyman BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods, 5(5) 409-415 (2008)
PDF
DOI
The interpretation of genome sequences requires reliable and standardized methods to assess protein function at high throughput. Here we describe a fast and reliable pipeline to study protein function in mammalian cells based on protein tagging in bacterial artificial chromosomes (BACs). The large size of the BAC transgenes ensures the presence of most, if not all, regulatory elements and results in expression that closely matches that of the endogenous gene. We show that BAC transgenes can be rapidly and reliably generated using 96-well-format recombineering. After stable transfection of these transgenes into human tissue culture cells or mouse embryonic stem cells, the localization, protein-protein and/or protein-DNA interactions of the tagged protein are studied using generic, tag-based assays. The same high-throughput approach will be generally applicable to other model systems.
2006
Mihail Sarov, Susan Schneider, Andrei I. Pozniakovsky, Assen Roguev, Susanne Ernst, Youming Zhang, Anthony A. Hyman, A. Francis Stewart A recombineering pipeline for functional genomics applied to Caenorhabditis elegans. Nat Methods, 3(10) 839-844 (2006)
PDF
DOI
We present a new concept in DNA engineering based on a pipeline of serial recombineering steps in liquid culture. This approach is fast, straightforward and facilitates simultaneous processing of multiple samples in parallel. We validated the approach by generating green fluorescent protein (GFP)-tagged transgenes from Caenorhabditis briggsae genomic clones in a multistep pipeline that takes only 4 d. The transgenes were engineered with minimal disturbance to the natural genomic context so that the correct level and pattern of expression will be secured after transgenesis. An example transgene for the C. briggsae ortholog of lin-59 was used for ballistic transformation in Caenorhabditis elegans. We show that the cross-species transgene is correctly expressed and rescues RNA interference (RNAi)-mediated knockdown of the endogenous C. elegans gene. The strategy that we describe adapts the power of recombineering in Escherichia coli for fluent DNA engineering to a format that can be directly scaled up for genomic projects.
Junping Wang, Mihail Sarov, Jeanette Rientjes, Jun Fu, Heike Hollak, Harald Kranz, Wei Xie, A Francis Stewart, Youming Zhang An improved recombineering approach by adding RecA to lambda Red recombination Mol Biotechnol, 32(1) 43-53 (2006)
PDF
Recombineering is the use of homologous recombination in Escherichia coli for DNA engineering. Of several approaches, use of the lambda phage Red operon is emerging as the most reliable and flexible. The Red operon includes three components: Redalpha, a 5' to 3' exonuclease, Redbeta, an annealing protein, and Redgamma, an inhibitor of the major E. coli exonuclease and recombination complex, RecBCD. Most E. coli cloning hosts are recA deficient to eliminate recombination and therefore enhance the stability of cloned DNAs. However, loss of RecA also impairs general cellular integrity. Here we report that transient RecA co-expression enhances the total number of successful recombinations in bacterial artificial chromosomes (BACs), mostly because the E. coli host is more able to survive the stresses of DNA transformation procedures. We combined this practical improvement with the advantages of a temperature-sensitive version of the low copy pSC101 plasmid to develop a protocol that is convenient and more efficient than any recombineering procedure, for use of either double- or single-stranded DNA, published to date.
2005
Mihail Sarov, A Francis Stewart The best control for the specificity of RNAi Trends Biotechnol, 23(9) 446-448 (2005)
PDF
DOI
RNA interference (RNAi) is revolutionizing functional genomics. However, there are several reasons to be concerned about the specificity and off-target effects of this technique. A recent paper by Kittler et al. describes a straightforward way to validate RNAi specificity, which exploits the increasing availability of bacterial artificial chromosome (BAC) clone resources. Genetic rescue of the RNAi phenotype by BAC transgenesis is the best control yet described for specificity, and has further implications for reverse genetics.
2004
Evdokia Pasheva, Mihail Sarov, Kiril Bidjekov, Iva Ugrinova, Bettina Sarg, Herbert Lindner, Iliya G Pashev In vitro acetylation of HMGB-1 and -2 proteins by CBP: the role of the acidic tail Biochemistry, 43(10) 2935-2940 (2004)
DOI
Histone acetyltransferases CBP, PCAF, and Tip60 have been tested for their ability to in vitro acetylate HMGB-1 and -2 proteins and their truncated forms lacking the C-terminal tail. It was found that these proteins were substrates for CBP only. Analyses of modified proteins by electrophoresis, amino acid sequencing, and mass spectrometry showed that full-length HMGB-1 and -2 were monoacetylated at Lys2. Removal of the C terminus resulted in (i) an increased incorporation of radiolabeled acetate within the proteins to a level close to that observed with histones H3/H4 and (ii) creation of a novel target site at Lys81. Acetylated and nonmodified HMGB-1 and -2 protein lacking the acidic tail were compared relative to their binding affinity to distorted DNA and the ability to bend linear DNA. Both proteins showed similar affinities to cisplatin-damaged DNA; the acetylated protein, however, was 3-fold more effective in inducing ligase-mediated circularization of a 111-bp DNA fragment. The alterations in the acetylation pattern of HMGB-1 and -2 upon removal of the C-terminal tail are regarded as a means by which the acidic domain modulates some properties of these proteins.
2003
Daniel Schaft*, Assen Roguev*, Kimberly M. Kotovic, Anna Shevchenko, Mihail Sarov, Andrej Shevchenko, Karla M. Neugebauer, A. Francis Stewart The histone 3 lysine 36 methyltransferase, SET2, is involved in transcriptional elongation. Nucleic Acids Res, 31(10) 2475-2482 (2003)
PDF
Existing evidence indicates that SET2, the histone 3 lysine 36 methyltransferase of Saccharomyces cerevisiae, is a transcriptional repressor. Here we show by five main lines of evidence that SET2 is involved in transcriptional elongation. First, most, if not all, subunits of the RNAP II holoenzyme co-purify with SET2. Second, all of the co-purifying RNAP II subunit, RPO21, was phosphorylated at serines 5 and 2 of the C-terminal domain (CTD) tail, indicating that the SET2 association is specific to either the elongating or SSN3 repressed forms (or both) of RNAP II. Third, the association of SET2 with CTD phosphorylated RPO21 remained in the absence of ssn3. Fourth, in the absence of ssn3, mRNA production from gal1 required SET2. Fifth, SET2 was detected on gal1 by in vivo crosslinking after, but not before, the induction of transcription. Similarly, SET2 physically associated with the transcribed region of pdr5 but was not detected on gal1 or pdr5 promoter regions. Since SET2 is also a histone methyltransferase, these results suggest a role for histone 3 lysine 36 methylation in transcriptional elongation.