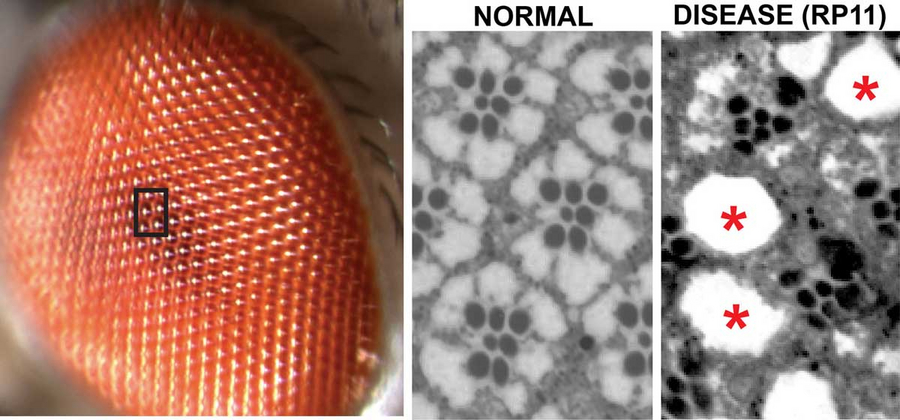
Image of the compound eye of Drosophila melanogaster with multiple repeated units of retinal cells. Black box is magnified in the microscopic images on the right, showing sections of a normal and of a degenerative (RP11 disease) fly eye. Loss of retinal cells is evident by holes (*) and disorganized units. © Hebbar et al. / MPI-CBG
Retinitis Pigmentosa (RP) is a hereditary neurodegenerative disease of the retina affecting 1 in 3.000 – 8.000 people worldwide, which leads to blindness due to the death of photoreceptor cells. The disease is characterized by an early onset of night blindness, followed by the loss of the peripheral vision and ultimately blindness. To date, mutations in more than 40 genes have been linked to RP.
Fruit fly as a model for human diseases
The research group of Elisabeth Knust at the Max Planck Institute of Molecular Cell Biology and Genetics in Dresden focusses on identifying mechanisms that underlie hereditary retinal degeneration. Together with Sarita Hebbar, a postdoctoral researcher in the lab and Malte Lehmann, a former member of the lab and currently a post-doctoral researcher and physician at the Charité in Berlin, the group has developed a model of RP11 in the fruit fly Drosophila melanogaster with defined mutations in the gene Prpf31. Mutations in the corresponding human gene are known to be linked with RP11. The researchers published their study in the journal “Biology Open.” The authors explain: “Flies carrying a mutation in this gene become blind. This is striking, given that the fly retina and the human retina are structurally diverse. However, their building blocks and many genes required for function are conserved in evolution. By modeling diseases in flies, questions of disease onset and progression with age and their cellular basis can be addressed in a living organism, which is not possible in cultured cells.”
Cutting genetic material into pieces
The gene Prpf31 is expressed in many tissues in our body and plays a role in generating appropriately sized messenger RNAs. mRNAs contain the instruction for the generation of proteins, which carry out the diverse functions in a cell. The process of cutting a longer RNA into smaller pieces and joining these together to form coding mRNAs is called splicing, and Prpf31 is part of the cellular splicing machinery. The authors add: “A disruption of splicing will result in mRNAs that encode proteins with no or defective functions. In our study we describe the generation of flies with defined mutations in Prpf31. These mutations mimic the retinal degeneration associated with RP11 in humans. This allowed us to identify a connection between mutation in Prpf31 and the cellular consequences leading to retinal degeneration. One of these consequences is an increased level of rhodopsin, the light-sensitive protein, which is instrumental for vision in both flies and human.” Strikingly, experimentally lowering the levels of rhodopsin by raising animals in food lacking vitamin A, which is required for the generation of functional rhodopsin, prevents retinal degeneration in mutant flies.”
This study opens avenues for future research. The model of the fruit fly can be used to investigate the general roles of Prpf31, for instance in tissues other than the eye. It may also help researchers to understand why the eye is especially vulnerable to splicing defects. Elisabeth Knust, the supervisor adds: “Another possibility is to further investigate how rhodopsin synthesis is regulated, since several human retinal diseases are associated with perturbations in rhodopsin synthesis or transport.”
Sarita Hebbar, Malte Lehmann, Sarah Behrens, Catrin Hälsig, Weihua Leng, Michaela Yuan, Sylke Winkler, Elisabeth Knust: Mutations in the splicing regulator Prp31 lead to retinal degeneration in Drosophila. Biology Open 2021 doi: 10.1242/bio.052332, Published 25 January 2021