Flexible Virtual Reality Visualization on the Java VM
We pioneered the use of Virtual Reality (VR) technology for volumetric 3D microscopy. We developed the open-source VR software framework scenery, which enables users to view terabyte-sized dynamic 3D+time microscopy datasets immersively. They can literally “walk inside” the specimen and observe the biological dynamics of tissue self-organization room-sized. This works both for previously recorded imagery and for live views during a running acquisition. The system transparently partitions the data onto multiple graphics processing units (GPUs) to achieve the multi-gigapixel/second rendering performance required for smooth and immediate immersion.
In collaboration with the Tomancak Lab (MPI-CBG), the Computer Graphics Lab of Prof. Stefan Gumhold (TU Dresden), and Kyle Harrington (MDC Berlin).
U. Günther, T. Pietzsch, A. Gupta, K. I. S. Harrington, P. Tomancak, S. Gumhold, and I. F. Sbalzarini. scenery: Flexible Virtual Reality Visualization on the Java VM. In Proc. IEEE Conference on Visualization, pages 166-170, Vancouver, Canada, 2019.
Self-organized shape dynamics of active surfaces
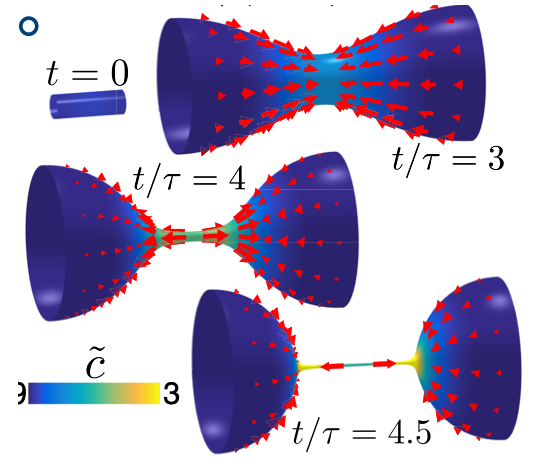
We numerically predicted the self-organized shape dynamics of active surfaces with tubular or spherical topology. This captures the physics of common morphologies in early organ or organoid development, such as spherical lumens, cysts, or tubular canaliculi. Shape changes in such tissues are driven by active mechanical forces that are generated by chemical processes, which in turn are affected by the deformations and flows. We integrated the interactions between the geometry of a deforming tissue and the active processes within by simulating the self-organized shape dynamics of active surfaces. We found that the tight coupling between surface mechanics and active processes gives rise to the spontaneous emergence and maintenance of polarity, cleavage, and directed peristaltic motion.
Learning physically consistent differential equation models from data using group sparsity
Biological processes are dynamic, nonlinear, and often involve yet unknown molecular interactions. There is hence great potential for data-driven machine learning of models, complementing traditional models derived from physical principles. Data-driven models can help identify values of unknown diffusion or rate constants, find minimal mechanisms, and enable mathematical analysis of systems for which first-principles modes are not (yet) known. But data-driven models often violate physical laws. As we show here, this can be remedied by constraining data-driven inference algorithms by prior knowledge from physics, such as conservation laws or symmetries in the molecular interactions. The resulting inference algorithm is shown to robustly learn physically consistent and interpretable models, even in cases where this fails without the physical prior.
In collaboration with the Müller Lab (LMU Munich & Flatiron CCM, New York).
Dominik Sturm, Ivo F. Sbalzarini Robust variable selection for spatial point processes observed with noise. Spatial Statistics, 74 Art. No. 101005 (2026)
Open AccessPDF
DOI
We propose a method for variable selection in the intensity function of spatial point processes that combines sparsity-promoting estimation with noise-robust model selection. As high-resolution spatial data becomes increasingly available through remote sensing and automated image analysis, identifying spatial covariates that influence the localization of events is crucial to understand the underlying mechanism. However, results from automated acquisition techniques are often noisy, for example due to measurement uncertainties and detection errors. We study the impact of such noise on sparse point-process estimation across different models. To improve noise robustness without requiring additional knowledge about the true process, we propose to use stability selection based on point-process subsampling and to incorporate a non-convex best-subset penalty to enhance sparsity. In extensive simulations, we demonstrate that this approach reliably recovers true covariates under diverse noise scenarios and improves both selection accuracy and stability. We then apply the proposed method to a forestry data set, analyzing the distribution of trees in a tropical rain forest. This shows the practical utility of the method for robust variable selection in spatial point-process models under noise, without requiring additional knowledge of the process.
Dominik Sturm*, Hiba Bensalem*, Ivo F. Sbalzarini Spatially Informed Autoencoders for Interpretable Visual Representation Learning.
In: International Conference on Learning Representations (ICLR)
(2026), Appleton WI, ICLR (2026), 1-35
Open AccessPDF
DOI
We introduce spatially informed variational autoencoders (SI-VAE) as self-supervised deep-learning models that use stochastic point processes to predict spatial organization patterns from images. Existing approaches to learning visual representations based on variational autoencoders (VAE) struggle to capture spatial correlations between objects or events, focusing instead on pixel intensities. We address this limitation by incorporating a point-process likelihood, derived from the Papangelou conditional intensity, as a self-supervision target. This results in a hybrid model that learns statistically interpretable representations of spatial localization patterns and enables zero-shot conditional simulation directly from images. Experiments with synthetic images show that SI-VAE improve the classification accuracy of attractive, repulsive, and uncorrelated point patterns from 48% (VAE) to over 80% in the worst case and 90% in the best case, while generalizing to unseen data. We apply SI-VAE to a real-world microscopy data set, demonstrating its use for studying the spatial organization of proteins in human cells and for using the representations in downstream statistical analysis.
Michael Hecht, Phil-Alexander Hofmann, Damar Wicaksono, Uwe Hernandez Acosta, Krzysztof Gonciarz, Jannik Kissinger, Vladimir Sivkin, Ivo F. Sbalzarini Multivariate Newton interpolation in downward closed spaces reaches the optimal Bernstein-Walsh approximation rate. IMA Journal of Numerical Anaysis, Art. No. doi: 10.1093/imanum/draf137 (2026)
Open Access DOI
Recent advances in Bernstein-Walsh theory have extended Bernstein's Theorem to multiple dimensions, stating that a multivariate function can be approximated with a geometric rate in a downward-closed polynomial space if and only if it is analytic in a generalized Bernstein polyellipse. To compute approximations of this class of functions-which we term Bos-Levenberg-Trefethen-(BLT) functions-we extend the classic univariate Newton interpolation algorithm to arbitrary multivariate downward-closed polynomial spaces, while maintaining its quadratic runtime and linear storage complexity. The present generalization supports any choice of (nontensorial) unisolvent interpolation nodes, whose number coincides with the dimension of the chosen downward-closed space. We prove that by selecting Leja nodes, the optimal geometric approximation rates for BLT-functions are achieved and that these rates extend to the derivatives of the interpolants. Choosing a Euclidean degree results in downward-closed spaces whose dimension only grows sub-exponentially with spatial dimension, while delivering approximation rates close to or matching those of the tensorial maximum-degree case, hence mitigating the curse of dimensionality. Importantly, our constructive proof directly inspires an algorithm for multivariate polynomial interpolation. We implemented this algorithm as a Python package and use it here to validate our theoretical findings in numerical experiments. These experiments corroborate the superiority of multivariate Newton interpolation over state-of-the-art alternatives, and they suggest that Leja-ordered Chebyshev-Lobatto nodes offer the same approximation power as Leja nodes.
Giovanni Volpe, Carolina Wählby, Lei Tian, Michael Hecht, Artur Yakimovich, Kristina Monakhova, Laura Waller, Ivo F. Sbalzarini, Christopher A Metzler, Mingyang Xie, Kevin Zhang, Isaac C D Lenton, Halina Rubinsztein-Dunlop, Daniel Brunner, Bijie Bai, Aydogan Ozcan, Daniel Midtvedt, Hao Wang, Nataša Sladoje, Joakim Lindblad, Jason T Smith, Marien Ochoa, Margarida Barroso, Xavier Intes, Tong Qiu, Li-Yu Yu, Sixian You, Yongtao Liu, Maxim A Ziatdinov, Sergei V Kalinin, Arlo Sheridan, Uri Manor, Elias Nehme, Ofri Goldenberg, Yoav Shechtman, Henrik K Moberg, Christoph Langhammer, Barbora Špačková, Saga Helgadottir, Benjamin Midtvedt, Aykut Argun, Tobias Thalheim, Frank Cichos, Stefano Bo, Lars Hubatsch, Jesus Pineda, Carlo Manzo, Harshith Bachimanchi, Erik Selander, Antoni Homs-Corbera, Martin Fränzl, Kevin de Haan, Yair Rivenson, Zofia Korczak, Caroline Beck Adiels, Mite Mijalkov, Dániel Veréb, Yu-Wei Chang, Joana B Pereira, Damian Matuszewski, Gustaf Kylberg, Ida-Maria Sintorn, Juan C Caicedo, Beth A Cimini, Muyinatu A Lediju Bell, Bruno M Saraiva, Guillaume Jacquemet, Ricardo Henriques, Wei Ouyang, Trang Le, Estibaliz Gómez-de-Mariscal, Daniel Sage, Arrate Muñoz-Barrutia, Ebba Josefson Lindqvist, Johanna Bergman Roadmap on Deep Learning for Microscopy. J. Phys. Photonics, 8(1) Art. No. 012501 (2026)
Open AccessPDF
DOI
Through digital imaging, microscopy has evolved from primarily being a means for visual observation of life at the micro- and nano-scale, to a quantitative tool with ever-increasing resolution and throughput. Artificial intelligence, deep neural networks, and machine learning (ML) are all niche terms describing computational methods that have gained a pivotal role in microscopy-based research over the past decade. This Roadmap encompasses key aspects of how ML is applied to microscopy image data, with the aim of gaining scientific knowledge by improved image quality, automated detection, segmentation, classification and tracking of objects, and efficient merging of information from multiple imaging modalities. We aim to give the reader an overview of the key developments and an understanding of possibilities and limitations of ML for microscopy. It will be of interest to a wide cross-disciplinary audience in the physical sciences and life sciences.
Lennart Justin Schulze Mesh-free methods for the adaptive discretization and high-order approximation of dynamic surfaces.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2026)
Open Access
In the past decades, mesh-free particle methods have advanced steadily and amassed a rich body of literature. They have successfully demonstrated their ability to simulate complex physics in various fields of research ranging from applied engineering to biology. Remarkably, the mesh-free representation of curved, dynamic surfaces has received far less attention than the mesh-free approximation of differential operators. The most commonly used, truly mesh-free approach for tracking and quantifying curved surfaces is limited to lower-order accuracy, with its fundamental basis having been proposed in the nineties. In this thesis, we bring high-order geometric computing to mesh-free particle methods. We propose the particle closest-point method, which implicitly tracks and quantifies evolving curved surfaces in a narrow band around the surface without the need of any connected mesh. It represents surfaces as the zero level-set of high-order polynomials that are obtained in local regression problems in a Newton-Lagrange basis on a set of unisolvent nodes, enabling robustness on scattered data. It is a purely geometric approach that computes comprehensive surface information in the form of signed-distance values, closest points, surface normals, and mean and Gaussian curvatures. We demonstrate that the surface quantities converge with the theoretical orders and show its capabilities in simulations of multi-phase hydrodynamics, where we consider oscillating and dividing droplets. We leverage the comprehensive and accurate geometric information of the particle closest-point method to address an issue that is encountered in solving partial differential equations on curved surfaces that are represented explicitly: When the surface undergoes large deformations, the surface point distribution inevitably becomes locally irregular, leading to a loss of accuracy and stability in the approximation of surface differential operators. We propose the self-adaptive implicit surface sampling method, which computes globally adaptive and locally regular point distributions for curved surfaces. It is based on two key mechanisms, the relaxation of the surface point distribution and a routine to find an appropriate number of points. The relaxation is done by minimizing a global potential which consists of local point-point interactions that depend on the local curvature of the surface. We minimize the global potential using a gradient descent accelerated by a line search. For finding an appropriate number of points for the discretization of the surface we propose a local integral support measure to decide if there is a lack or excess of points. We show the potential of the proposed method by computing adaptive and locally regular point discretizations of a variety of parametric and non-parametric surfaces from both synthetic and real-world data. We find robust and rapid convergence to the final point set size based on the integral support measure and low average deviations from the target spacing of the points. To demonstrate the impact of the contributions of this thesis we combine the two methods with the surface variant of discretization-corrected particle strength exchange to solve partial differential equations on deformable surfaces and coupled bulk-surface equations. We propose time-integration schemes that outline how the methods can be combined. We solve the reaction-diffusion equation on the surface of an oscillating droplet and show various Turing patterns. We further simulate a morphogenetic model, where local surface concentrations induce growth of the surface. We reveal the importance of maintaining a regular surface discretization by means of the self-adaptive implicit surface sampling method and find rich, organic shapes governed by the morphogenetic model.
Abhinav Singh, Landfried Kraatz, Serhii Yaskovets, Pietro Incardona, Ivo F. Sbalzarini Integrating Odeint Time Stepping into OpenFPM for Distributed and GPU Accelerated Numerical Solvers Journal of Open Research Software, 14(1) Art. No. 15 (2026)
Open AccessPDF
DOI
We present a scientific numerical software for multi-stage, multi-step, and adaptive explicit time integration on distributed-memory parallel computers and on Graphics Processing Units (GPUs). Our implementation integrates the Odeint library from Boost with the OpenFPM framework for scientific computing, enabling compact and scalable numerical simulation codes. Specifically, we extend the Odeint data types to OpenFPM’s metaprogramming system. This makes the time-integration methods from Odeint available in a concise template-expression language for numerical codes distributed and parallelized using OpenFPM. We benchmark the software for exponential and sigmoidal dynamics and present application examples to the 3D Gray-Scott reaction-diffusion problem and the “dam break” problem from fluid mechanics. We find a strong-scaling efficiency of 80% on up to 512 CPU cores and a five-fold speedup on a single GPU.
Roua Rouatbi, Juan-Esteban Suarez Cardona, Alba Villaronga Luque, Jesse V Veenvliet, Ivo F. Sbalzarini A Continuous and Interpretable Morphometric for Robust Quantification of Dynamic Biological Shapes
In: Proc. IEEE Intl. Symposium Biomedical Imaging (ISBI)
(2026), Piscataway, N.J., IEEE (2026), 1-5
PDF
DOI
We introduce the Push-Forward Signed Distance Morphometric (PF-SDM) for shape quantification in biomedical imaging. The PF-SDM compactly encodes geometric and topological properties of closed shapes, including their skeleton and symmetries. This provides robust and interpretable features for shape comparison and machine learning. The PF-SDM is mathematically smooth, providing access to gradients and differential-geometric quantities. It also extends to temporal dynamics and allows fusing spatial intensity distributions, such as genetic markers, with shape dynamics. We present the PF-SDM theory, benchmark it on synthetic data, and apply it to predicting body-axis formation in mouse gastruloids, outperforming a CNN baseline in both accuracy and speed.
2025
Roua Rouatbi, Juan-Esteban Suarez Cardona, Alba Villaronga-Luque, Jesse V Veenvliet, Ivo F. Sbalzarini A Continuous and Interpretable Morphometric for Robust Quantification of Dynamic Biological Shapes. ArXiv, Art. No. ArXiv:2410.21004 (2025)
Open Access
We introduce the Push-Forward Signed Distance Morphometric (PF-SDM) for shape quantification in biomedical imaging. The PF-SDM compactly encodes geometric and topological properties of closed shapes, including their skeleton and symmetries. This provides robust and interpretable features for shape comparison and machine learning. The PF-SDM is mathematically smooth, providing access to gradients and differential-geometric quantities. It also extends to temporal dynamics and allows fusing spatial intensity distributions, such as genetic markers, with shape dynamics. We present the PF-SDM theory, benchmark it on synthetic data, and apply it to predicting body-axis formation in mouse gastruloids, outperforming a CNN baseline in both accuracy and speed.
Max Rosenkranz, Markus Kaestner, Ivo F. Sbalzarini Data-Efficient Inverse Design of Spinodoid Metamaterials. Integrating materials and manufacturing innovation, Art. No. doi: 10.1007/s40192-025-00426-1 (2025)
Open Access DOI
We present a data-efficient neural-network model for predicting linear-elastic properties of spinodoid metamaterials from their mesoscale structure. Our machine-learning model requires 75 data points for training, greatly improving data efficiency over previous models that required thousands of training samples. We achieve this by leveraging concepts from geometric learning. Specifically, we exploit physical properties, such as positive semi-definiteness of the elasticity tensor, as well as structural invariances and equivariances of the problem, for example with respect to coordinate axes permutations. The neural network model is designed to exactly fulfill these constraints; it does not have to learn them from data. The resulting model enables data- and compute-efficient inverse design of spinodoid metamaterials. In inverse design, the goal is to find a material mesostructure that leads to desired mechanical properties on the macroscale. Exactly fulfilling physical and structural constraints, the present neural network model remains differentiable. This allows using fast gradient-based optimizers for inverse design. We demonstrate this by inversely designing spinodoid metamaterials that achieve desired linear elastic target properties in three dimensions. Inverse design is treated as a constrained optimization problem over the parameters describing the metamaterial. The results confirm that the present approach requires significantly less training data than previous machine-learning approaches and allows incorporating multiple objectives in the inverse design process. Since the structure of the design space is independent of the target material properties, we hope that such data-efficient models will be useful also for inverse design of spinodoids beyond linear elasticity.
Camilo A. S. Afanador*, Stéphane Urcun*, Ivo F. Sbalzarini, Stéphane P. A. Bordas, Olga Barrera, Mohammad Mahdi Rajabi, Romain Seil, Anas Obeidat Mechanics of knee meniscus results from precise balance between material microstructure and synovial fluid viscosity. PLoS ONE, 20(9) Art. No. e0304440 (2025)
Open AccessPDF
DOI
The meniscus plays a crucial role in the biomechanics of the knee, serving as load transmitter and reducing friction between joints. Understanding the biomechanics of the meniscus is essential to effective treatment of knee injuries and degenerative conditions. This study aims to elucidate the relationship between the porous microstructure of the human knee meniscus and its biomechanical function, specifically focusing on fluid dynamics at the pore scale. Here, we use two central-meniscus samples extracted from a human knee and reconstruct high-resolution geometry models from [Formula: see text]-CT scans. By eroding the channels of the original meniscus geometry, we simulate perturbed microstructures with varying porosities ( 53% to 80%), whilst preserving the connectivity of the porous structure. We numerically solve for the fluid dynamics in the meniscus using a mesh-free particle method, considering various inlet pressure conditions, characterising the fluid flow within the microstructures. The results of the original microstructure associated with a physiological dynamic viscosity of synovial fluid are in accordance with biophysical experiments on menisci. Furthermore, the eroded microstructure with a 33% increase in porosity exhibited a remarkable 120% increase in flow velocity. This emphasises the sensitivity of meniscus physiology to the porous microstructure, showing that detailed computational models can explore physiological and pathological conditions, advancing further knee biomechanics research.
Johannes Pahlke, Ivo F. Sbalzarini On the Computational Power of Particle Methods Fundamenta Informaticae, 194(1) Art. No. 3 (2025)
Open AccessPDF
DOI
We investigate the computational power of particle methods, a well-established class of algorit hms with applications in scientific computing and computer simulation. The computational power of a compute model determines the class of problems it can solve. Automata theory allows describing the computational power of abstract machines (automata) and the problems they can solve. At the top of the Chomsky hierarchy of formal languages and grammars are Turing machines, which resemble the concept on which most modern computers are built. Although particle methods can be interpreted as automata based on their formal definition, their computational power has so far not been studied. We address this by analyzing Turing completeness of particle methods. In particular, we prove two sets of restrictions under which a particle method is still Turing powerful, and we show when it loses Turing powerfulness. This contributes to understanding the theoretical foundations of particle methods and provides insight into the powerfulness of computer simulations.
Justina Stark, Ivo F. Sbalzarini#, Michael Brand# The people behind the papers - Justina Stark, Ivo Sbalzarini and Michael Brand. Development, 152(13) Art. No. dev205004 (2025)
DOI
Morphogens are secreted from a local source and form long-range gradients. Existing computational models to study morphogen gradients typically simplify the tissue geometry. In a new study, Justina Stark and colleagues investigate the contribution of the porous 3D tissue geometry to long-range Fgf8a gradients in the early zebrafish embryo. To learn more about the people behind this work, we caught up with first author Justina Stark, and corresponding authors Ivo Sbalzarini (Professor at TU Dresden and Group Leader at the Max Planck Institute of Molecular Cell Biology and Genetics) and Michael Brand (Professor at the Center for Regenerative Therapies, TU Dresden).
Justina Stark#, Rohit Krishnan Harish, Ivo F. Sbalzarini, Michael Brand# Morphogen gradients are regulated by porous media characteristics of the developing tissue. Development, 152(13) Art. No. dev204312 (2025)
Open Access DOI
Long-range morphogen gradients have been proposed to form by morphogen diffusion from a localized source to distributed sinks in the target tissue. The role of the complex tissue geometry in this process is, however, less well understood and has not been explicitly resolved in existing models. Here, we numerically reconstruct pore-scale 3D geometries of zebrafish epiboly from light-sheet microscopy volumes. In these high-resolution 3D geometries, we simulate Fgf8a gradient formation in the tortuous extracellular space. Our simulations show that when realistic embryo geometries are considered, a source-diffusion-degradation mechanism with additional binding to extracellular matrix polymers is sufficient to explain emergence and robust maintenance of Fgf8a gradients. The predicted normalized gradient is robust against changes in source and sink rates but sensitive to changes in the pore connectivity of the extracellular space, with lower connectivity leading to steeper and shorter gradients. This demonstrates the importance of considering realistic geometries when studying morphogen gradients.
Meri Abgaryan*, Xinning Cui*, Nandu Gopan, Gabriel della Maggiora, Artur Yakimovich, Ivo F. Sbalzarini Regularized Gradient Statistics Improve Generative Deep Learning Models of Super Resolution Microscopy. Small Methods, 9 Art. No. 202401900(1-14) (2025)
Open Access DOI
It is shown that regularizing the signal gradient statistics during training of deep-learning models of super-resolution fluorescence microscopy improves the generated images. Specifically, regularizing the images in the training data set is proposed to have gradient and Laplacian statistics closer to those expected for natural-scene images. The BioSR data set of matched pairs of diffraction-limited and super-resolution images is used to evaluate the proposed regularization in a state-of-the-art generative deep-learning model of super-resolution microscopy, the Conditional Variational Diffusion Model (CVDM). Since the proposed regularization is applied as a preprocessing step to the training data, it can be used in conjunction with any supervised machine-learning model. However, its utility is limited to images for which the prior is appropriate, which in the BioSR data set are the images of filamentous structures. The quality and generalization power of CVDM trained with and without the proposed regularization are compared, showing that the new prior yields images with clearer visual detail and better small-scale structure.
Paul Hempel*, Aryaman Gupta*, Ivo F. Sbalzarini, Stefan Gumhold A Transparent and Efficient Extension of IceT for Parallel Compositing on Non-Convex Volume Domain Decompositions.
In: Proceedings Eurographics Symposium on Parallel Graphics and Visualization (EGPGV)
(2025), The Eurographics Association (2025), 1-5
Open AccessPDF
DOI
The IceT library is widely used for parallel compositing but does not support non-convex volume domain decompositions. We provide a backward-compatible extension of IceT to handle non-convex domain decompositions of volume data. These are frequently produced in numerical simulations, but it is challenging to render them in parallel due to the non-commutativity of alpha compositing. We enable parallel volume rendering of non-convex domains in IceT by extending its parallel compositing to layered images. Our code follows an embedded design, extending and generalizing IceT's internal functions for image compression, splitting, compositing, and decompression to efficiently handle layered images, while maintaining the existing functionality and API. We perform scalability tests and provide our implementation open-source in a public repository, with in-line documentation and integration tests.
Alejandra Maria Foggia Numerical solution of scalar and vector differential equations on surface point clouds.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2025)
Open Access
In this thesis, I present the Surface Discretization-Corrected Particle Strength Exchange (Surface DC-PSE) method for vector-valued partial differential equations (PDEs) on curved surfaces. Surface DC-PSE is a numerical meshfree collocation method for approximating surface differential operators. The method requires a surface point cloud and the surface normal at each point, and provides a mathematically embedding but computationally embedding-free approximation of the fields and their spatial derivatives at each point. The Surface DC-PSE method is an extension of the DC-PSE method, which is a mesh- free numerical method for approximating Euclidean differential operators. Surface DC-PSE is based on the idea of extending the field constant along the normal into the embedding space, constructing the kernels using both surface and “virtual” extended particles along the normal, and collapsing the resulting ”bulk” kernels into surface ones. This thesis presents a comprehensive study of the Surface DC-PSE method, including its mathematical formulation, numerical implementation, and verification tests. The results show that the method provides convergent and stable approximations of scalar and vector surface differential operators, and is applicable to a wide range of surfaces. I also discuss the limitations of the method, including the requirement of a homogeneous point cloud, the sensitivity to particle distributions, and the computational cost of constructing the kernels. Future work is proposed to address these limitations, including modifying the neighborhood construction algorithm, evaluating the accuracy and stability of vector Surface DC-PSE on deforming surfaces, and exploring applications of the method to non-analytical surfaces. The Surface DC-PSE method has the potential to be used in a wide range of applications, including simulating biological fluid surfaces, studying morphogenesis, and modeling complex surface phenomena. The method provides a robust and reliable framework for approximating surface differential operators, and it provides a basis for further research and development in these and other areas of scientific computing.
Joel Jonsson, Bevan Cheeseman, Ivo F. Sbalzarini APR-CNN: Convolutional Neural Networks for the Adaptive Particle Representation of Large Microscopy Images. Transactions on Machine Learning Research, Art. No. 5qKI2dkrjL (2025)
Open AccessPDF
We present APR-CNN, a novel class of convolutional neural networks designed for efficient and scalable three-dimensional microscopy image analysis. APR-CNNs operate natively on a sparse, multi-resolution image representation known as the Adaptive Particle Representation (APR). This significantly reduces memory and compute requirements compared to traditional pixel-based CNNs. We introduce APR-native layers for convolution, pooling, and upsampling, along with hybrid architectures that combine APR and pixel layers to balance accuracy and computational efficiency. We show in benchmarks that APR-CNNs achieve comparable segmentation accuracy to pixel-based CNNs while drastically reducing memory usage and inference time. We further showcase the potential of APR-CNNs in large-scale volumetric image analysis, reducing inference times from weeks to days. This opens up new avenues for applying deep learning to large, high-resolution, three-dimensional biomedical datasets with constrained computational resources.
2024
Tina Subic, Ivo F. Sbalzarini Loss of bimolecular reactions in reaction-diffusion master equations is consistent with diffusion limited reaction kinetics in the mean field limit. J Chem Phys, 161(23) Art. No. 234107 (2024)
Open AccessPDF
DOI
We show that the resolution-dependent loss of bimolecular reactions in spatiotemporal Reaction-Diffusion Master Equations (RDMEs) is in agreement with the mean-field Collins-Kimball (C-K) theory of diffusion-limited reaction kinetics. The RDME is a spatial generalization of the chemical master equation, which enables studying stochastic reaction dynamics in spatially heterogeneous systems. It uses a regular Cartesian grid to partition space into locally well-mixed reaction compartments and treats diffusion as a jump reaction between neighboring grid cells. As the chance for reactants to be in the same grid cell decreases for smaller cell widths, the RDME loses bimolecular reactions in finer grids. We show that for a single homo-bimolecular reaction, the mesh spacing can be interpreted as the reaction radius of a well-mixed C-K rate. Then, the bimolecular reaction loss is consistent with diffusion-limited kinetics in the mean-field steady state. In this interpretation, the constant in a bimolecular reaction propensity is no longer the macroscopic reaction rate but the rate of the ballistic C-K step. For the same grid resolution, different diffusion models in RDME, such as those based on finite differences and Gaussian jumps, represent different reaction radii.
Dominik Sturm, Suryanarayana Maddu, Ivo F. Sbalzarini Learning locally dominant force balances in active particle systems. Proc Roy Soc A, 480(2304) Art. No. 20230532 (2024)
Open AccessPDF
DOI
We use a combination of unsupervised clustering and sparsity-promoting inference algorithms to learn locally dominant force balances that explain macroscopic pattern formation in self-organized active particle systems. The self-organized emergence of macroscopic patterns from microscopic interactions between self-propelled particles can be widely observed in nature. Although hydrodynamic theories help us better understand the physical basis of this phenomenon, identifying a sufficient set of local interactions that shape, regulate and sustain self-organized structures in active particle systems remains challenging. We investigate a classic hydrodynamic model of self-propelled particles that produces a wide variety of patterns, such as asters and moving density bands. Our data-driven analysis shows that propagating bands are formed by local alignment interactions driven by density gradients, while steady-state asters are shaped by a mechanism of splay-induced negative compressibility arising from strong particle interactions. Our method also reveals analogous physical principles of pattern formation in a system where the speed of the particle is influenced by the local density. This demonstrates the ability of our method to reveal physical commonalities across models. The physical mechanisms inferred from the data are in excellent agreement with analytical scaling arguments and experimental observations.
Joel Jonsson Efficient content-adaptive processing of large-scale fluorescence microscopy data.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2024)
Open Access
Fluorescence microscopy is a pivotal technology in biological research, enabling high-resolution imaging of cellular and subcellular structures and processes. Modern imaging modalities, such as light-sheet microscopy, are able to acquire images with high spatial and temporal resolution over large areas or long durations, leading to the routine generation of terabyte-sized datasets. Diverse image processing algorithms are required to extract useful information from these image datasets, but their application is impeded by the vast data size. This “data bottleneck” often limits the throughput and scalability of fluorescence microscopy studies and leads to under-utilization of the information contained in the images. This thesis addresses the computational challenges of processing large image volumes by leveraging the Adaptive Particle Representation (APR). The APR is a multi-resolution image representation that optimally adapts the local sampling density to the image contents, thereby reducing redundancies in the representation of sparse images typical of fluorescence microscopy. We build upon the APR and its previously existing software to enable a wide range of image processing methods, from basic filtering operations to advanced deep-learning techniques, to leverage the data-efficient APR format for enhanced computational efficiency and greatly reduced memory requirements on parallel computer architectures. We demonstrate in real large-scale imaging applications that this can provide a comprehensive solution to the data bottleneck in fluorescence microscopy. First, we present data structures and algorithms that enable efficient and native processing on APR images using multi-core CPU and GPU parallelization. We define an adaptation of discrete convolutions, which are essential for many image processing tasks, and strategies for defining scale-adaptive filters that exploit the varying spatial scales of the APR. We demonstrate the viability of this approach in the task of image deconvolution on synthetic and real images, and quantify the computational efficiency of our implementation compared to pixel convolutions on evenly sampled data. Second, we demonstrate the practical utility of APR-based image processing in large-scale neurohistology applications. We present methods that enable complete APR-native pipelines, including automatic APR conversion, multi-tile stitching, segmentation, visualization, and atlas registration. Applied in imaging experiments on an entire mouse brain and a large section of human brain tissue, our pipeline exhibits substantially increased efficiency over established voxel methods, achieving 115-fold reduced storage requirements and 71 times accelerated processing, enabling acquisition-rate processing on a modest workstation CPU. Finally, we adapt convolutional neural networks (CNNs) to operate natively on the APR, resulting in APR-CNNs that leverage the APR data structures to reduce their memory footprint and computational burden. Given that computationally intensive CNNs have emerged as the state of the art across a wide range of image processing tasks, this adaptation greatly broadens the applicability of APR-based processing. We evaluate the performance of APR-CNNs in instance segmentation on real microscopy data, showing that they can achieve comparable segmentation accuracy to traditional pixel CNNs despite significantly reduced input data size. This thesis demonstrates the potential of APR-native image processing as a transformative tool for fluorescence microscopy. By developing and optimizing data structures, algorithms, and pipelines tailored for data-efficient APR images, this work paves the way toward comprehensive solutions to the data bottleneck in large-scale imaging through a combination of data reduction and parallel processing. In particular, the adaptation of deep-learning methods has broad applicability, potentially leading to more efficient and scalable bioimaging workflows that can accelerate and reduce the cost of scientific discovery.
Charvi Jain, Sahar Vahdati, Nandu Gopan, Ivo F. Sbalzarini, Jens Lehmann Evaluating Large Language Model Literature Reviews In Interdisciplinary Science: A Systems Biology Perspective.
In: EKAW 2024: EKAW 2024 Workshops, Tutorials, Posters and Demos, 24th International Conference on Knowledge Engineering and Knowledge Management (EKAW 2024), November 26-28, 2024, Amsterdam, The Netherlands
(2024) Ch. 173, Aachen, CEUR Workshop Proceedings (2024), 1-6
Open AccessPDF
DOI
We evaluate the effectiveness of current large language model (LLM) literature review systems in interdisciplinary domains. While LLMs can support and accelerate reviewing the scientific literature, it is unclear how they cope with interdisciplinary science, where sources from multiple fields must be integrated according to relevance defined by context. We study this from the perspective of systems biology, a field that combines biology, mathematics, physics, and computer science. Using a set of expert-defined research questions, we assess the ability of LLMs to meaningfully integrate cross-domain knowledge and correctly reflect relevance. Specifically, we evaluate the quality of generated reports and the relevance of retrieved references from five different review models. We find that LLMs are a valuable augmentative tool for literature reviews, but trade off report quality for completeness in interdisciplinary domains. We address these limitations by proposing a novel method, termed AURORA, which is particularly designed for interdisciplinary applications. On the interdisciplinary systems biology benchmark, AURORA offers good coverage with high-quality reports.
Johannes Pahlke, Ivo F. Sbalzarini Proven Distributed Memory Parallelization of Particle Methods ACM Trans. Parallel Comput., 11(4) Art. No. 17 (2024)
Open AccessPDF
DOI
We provide a mathematically proven parallelization scheme for particle methods on distributed-memory computer systems. Particle methods are a versatile and widely used class of algorithms for computer simulations and numerical predictions in various applications, ranging from continuum fluid dynamics to discrete granular flows and molecular dynamics simulations. Particle methods naturally lend themselves to implementation on parallel computing systems. So far, however, a mathematical proof of correctness and equivalence to sequential implementations has only been available for shared-memory parallelism. Here, we leverage a formal definition of the algorithmic class of particle methods to provide a proven parallelization scheme for distributed-memory computers. We prove that thus parallelized particle methods on distributed-memory computers are formally equivalent to their sequential counterpart for a well-defined class of particle methods, and we provide analytical expressions for the speedup and scalability bounds of this class of algorithms in function of their parameters. The parallelization scheme analyzed here is the basis of many real-world software designs for parallel particle methods. The present analysis is, therefore, of direct relevance to existing and new parallel implementations of particle methods and places them on solid theoretical grounds, rationalizing best practices and providing useful scalability and speedup bounds for benchmarking.
Lennart Justin Schulze, Sachin K. T. Veettil, Ivo F. Sbalzarini A high-order fully Lagrangian particle level-set method for dynamic surfaces. J Comput Phys, 515 Art. No. 113262 (2024)
Open AccessPDF
DOI
We present a fully Lagrangian particle level-set method based on high-order polynomial regression. This enables meshfree simulations of dynamic surfaces, relaxing the need for particle-mesh interpolation. Instead, we perform level-set redistancing directly on irregularly distributed particles by polynomial regression in a Newton-Lagrange basis on a set of unisolvent nodes. We demonstrate that the resulting particle closest-point (PCP) redistancing achieves high-order accuracy for 2D and 3D geometries discretized on irregular particle distributions and has better robustness against particle distortion than regression in a monomial basis. Further, we show convergence in classic level-set benchmark cases involving ill-conditioned particle distributions, and we present an example application to multi-phase flow problems involving oscillating and dividing droplets.
Salman Alam, Bibi Najma, Abhinav Singh, Jeremy Laprade, Gauri Gajeshwar, Hannah G. Yevick, Aparna Baskaran, Peter J Foster, Guillaume Duclos Active Freedericksz Transition in Active Nematic Droplets. Physical Review X, 14(4) Art. No. 041002 (2024)
Open Access DOI
Active nematic liquid crystals have the remarkable ability to spontaneously deform and flow in the absence of any external driving force. While living materials with orientational order, such as the mitotic spindle, can self-assemble in quiescent active phases, reconstituted active systems often display chaotic, periodic, or circulating flows under confinement. Quiescent in vitro active nematics are, therefore, quite rare, despite the prediction from active hydrodynamic theory that confinement between two parallel plates can suppress flows. This spontaneous flow transition-named the active Freedericksz transition by analogy with the conventional Freedericksz transition in passive nematic liquid crystals under a magnetic field-has been a cornerstone of the field of active matter. Here, we report experimental evidence that confinement in spherical droplets can stabilize the otherwise chaotic dynamics of a 3D extensile active nematics, giving rise to a quiescent-yet still out-of-equilibrium-nematic liquid crystal. The active nematics spontaneously flow when confined in larger droplets. The composite nature of our model system composed of extensile bundles of microtubules and molecular motors dispersed in a passive colloidal liquid crystal allows us to demonstrate how the interplay of activity, nematic elasticity, and confinement impacts the spontaneous flow transition. The critical diameter increases when motor concentration decreases or nematic elasticity increases. Experiments and simulations also demonstrate that the critical confinement depends on the confining geometry, with the critical diameter in droplets being larger than the critical width in channels. Biochemical assays reveal that neither confinement nor nematic elasticity impacts the energy-consumption rate, confirming that the quiescent active phase is the stable out-of-equilibrium phase predicted theoretically. Further experiments in dense arrays of monodisperse droplets show that fluctuations in the droplet composition can smooth the flow transition close to the critical diameter. In conclusion, our work provides experimental validation of the active Freedericksz transition in 3D active nematics, with potential applications in human health, ecology, and soft robotics.
Lennart Beck, Ivo F. Sbalzarini When Particle Shifting is Expected to Improve Convergence of Lagrangian Smoothed Particle Hydrodynamics.
In: Proceedings 18th SPHERIC International Workshop, 18-20 June 2024
(2024), Berlin, Germany, SPHERIC (2024), 289-296
PDF
DOI
We introduce a metric to determine when particle shifting is expected to improve the convergence of weakly compressible Lagrangian SPH. Shifting regularizes particle positions by biasing them toward a more homogeneous distribution. However, shifting also introduces an additional discretization error. To quantify these two opposing effects, we analyze the two- dimensional Taylor-Green case and show that particle shifting is advantageous when there is strong-enough distortion in the particle distribution. We propose a dimensionless metric of particle distortion to predict this break-even point, which we show to behave qualitatively similar across simulation cases. Since the proposed metric can be computed in a running simulation, it can be used to control the onset of shifting.
Suse Seidemann*, Florian Salomon*, Karl Hoffmann, Thomas Kurth, Ivo F. Sbalzarini, Robert Haase, Marius Ader Automated quantification of photoreceptor outer segments in developing and degenerating retinas on microscopy images across scales. Front Mol Neurosci, 17 Art. No. 1398447 (2024)
Open Access DOI
The functionality of photoreceptors, rods, and cones is highly dependent on their outer segments (POS), a cellular compartment containing highly organized membranous structures that generate biochemical signals from incident light. While POS formation and degeneration are qualitatively assessed on microscopy images, reliable methodology for quantitative analyses is still limited. Here, we developed methods to quantify POS (QuaPOS) maturation and quality on retinal sections using automated image analyses. POS formation was examined during the development and in adulthood of wild-type mice via light microscopy (LM) and transmission electron microscopy (TEM). To quantify the number, size, shape, and fluorescence intensity of POS, retinal cryosections were immunostained for the cone POS marker S-opsin. Fluorescence images were used to train the robust classifier QuaPOS-LM based on supervised machine learning for automated image segmentation. Characteristic features of segmentation results were extracted to quantify the maturation of cone POS. Subsequently, this quantification method was applied to characterize POS degeneration in "cone photoreceptor function loss 1" mice. TEM images were used to establish the ultrastructural quantification method QuaPOS-TEM for the alignment of POS membranes. Images were analyzed using a custom-written MATLAB code to extract the orientation of membranes from the image gradient and their alignment (coherency). This analysis was used to quantify the POS morphology of wild-type and two inherited retinal degeneration ("retinal degeneration 19" and "rhodopsin knock-out") mouse lines. Both automated analysis technologies provided robust characterization and quantification of POS based on LM or TEM images. Automated image segmentation by the classifier QuaPOS-LM and analysis of the orientation of membrane stacks by QuaPOS-TEM using fluorescent or TEM images allowed quantitative evaluation of POS formation and quality. The assessments showed an increase in POS number, volume, and membrane coherency during wild-type postnatal development, while a decrease in all three observables was detected in different retinal degeneration mouse models. All the code used for the presented analysis is open source, including example datasets to reproduce the findings. Hence, the QuaPOS quantification methods are useful for in-depth characterization of POS on retinal sections in developmental studies, for disease modeling, or after therapeutic interventions affecting photoreceptors.
Jan Tiemann#, Matthew McGinity, Ivo F. Sbalzarini, Ulrik Günther# Live and Interactive 3D Photomanipulation under the Microscope using Virtual Reality.
In: CHI'24 : extended abstracts of the 2024 CHI Conference on Human Factors in Computing Systems
(2024) Ch. 228(Eds.) Florian Mueller, New York, ACM (2024)
Open AccessPDF
DOI
State-of-the-art microscopes, as used in cell biology, are not only capable of capturing 3D images, but also permit manipulation of (sub-)cellular structures using techniques such as optical traps, optogenetics or laser ablation. However, such microscopes are still controlled using 2D interfaces, prohibiting actual 3-dimensional manipulation.
We present microscenery, a virtual reality (VR) microscope control software, designed to facilitate 3D laser ablation experiments. We combine microscopy automation with VR rendering and intuitive controller-based input to empower biologists with the precision of laser-based techniques while providing the full 3D spatial context of their sample. We describe the design goals and architecture of the software and illustrate the potential of the system by conducting a brief expert review study for 3D ablation experiments. Our results suggest VR is not only an effective interface for microscopic manipulations, but can enable novel experiments which are either impossible with traditional 2D interfaces, or prohibitively time-consuming.
Justina Stark Solving continuous reaction-diffusion models in image-based complex geometries.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2024)
Open Access
Porous media, including soil, catalysts, rocks, and organic tissue, are ubiquitous in nature, acting as complex environments through which heat, ions, and chemicals travel. Diffusion, often coupled to interfacial reactions, constitutes a fundamental transport process in porous media. It plays an important role in the transport of fertilizer and contaminants in soil, heat conduction in insulators, and natural phenomena such as geological rock transformations and biological signaling and patterning. This thesis aims to enable a deeper understanding of reaction-diffusion processes in porous media by developing a flexible and computationally efficient numerical modeling and simulation workflow. Numerical modeling is required whenever the problem is too complex for mechanistic insight by quantitative experiments or analytical theory. Reaction-diffusion processes in porous media are such a complex problem, as transport is coupled to the intricate pore geometry. In addition, they involve different scales, from microscale tortuous diffusion pathways and local reactions to macroscale gradients, requiring models that resolve multiple scales. Multiscale modeling is, however, challenging due to its large memory requirement and computational cost. In addition, realistic porous media geometries, as can be derived from microscopy images or µCTs, are not parametrizable, requiring algorithmic representation. We address these issues by developing a scalable, multi-GPU accelerated numerical simulation pipeline that enables memory-efficient multiscale modeling of reaction-diffusion processes in realistic, image-based geometries. This pipeline takes volumetric images as input, from which it derives implicit geometry representations using the level-set method. The diffusion domain is discretized in a geometry-adapted, memory-efficient way using distributed sparse block grids. Reaction-diffusion PDEs are solved in the strong form using the finite difference method with scalable multi-GPU acceleration, enabling the simulation in large, highly resolved 3D samples. We demonstrate the versatility of the present pipeline by simulating reaction-diffusion processes in the image-derived 3D geometries of four applications: fertilizer diffusion in soil, heat conduction with surface dissipation in reticulate porous ceramics, fluid-mediated mineral replacement in rocks, and morphogen gradient formation in the extracellular space of a gastrulating zebrafish embryo. The former two are used to benchmark the performance of our pipeline, whereas the latter two address real-world problems from geology and biology, respectively. The geological problem considers a process called dolomitization, which converts calcite into dolomite. Determining the geophysical characteristics of the earth's most abundant rocks, dolomitization plays an important role in engineering and geology. Predicting dolomitization is hampered by the extreme scales involved, as mountain-scale dolomite is produced by ion-scale reactions over millions of years. Using the presented pipeline, we derive rock geometries from µCTs and simulate dolomitization as an inhomogeneous reaction-diffusion process with moving reaction fronts and phase-dependent diffusion. The simulation results show that reaction and diffusion are not sufficient to explain the reaction-front roughness observed experimentally, implying that other processes, such as advection, porosity fingering, or sub-resolution geometric features, such as microcracks in the rock, play an important role in dolomitization. The biological problem, which constitutes the main application of this thesis, is the formation of morphogen gradients during embryonic development. This is a particularly complex problem influenced by several factors, such as dynamically changing tissue geometries, localized sources and sinks, and interaction with molecules of the extracellular matrix (e.g., HSPG). The abundance of factors involved and the coupling between them makes it difficult to quantify how they modulate the gradient individually and collectively. We use the present pipeline to reconstruct realistic extracellular space (ECS) geometries of a zebrafish embryo from a light-sheet microscopy video. In these geometries, we simulate the gradient formation of the morphogen Fgf8a, showing for the first time in realistic embryo geometries that a source-diffusion-degradation mechanism with HSPG binding is sufficient for the spontaneous formation and maintenance of robust long-range morphogen gradients. We further test gradient sensitivity against different source, sink, and HSPG-binding rates and show that the gradient becomes distorted when ECS volume or connectivity in the model changes, demonstrating the importance of considering realistic embryo geometries. In summary, this thesis shows that modeling highly resolved, realistic 3D geometries is computationally feasible using geometry-adapted sparse grids, achieving an 18-fold reduction in memory requirements for the zebrafish model compared to a dense-grid implementation. Multi-CPU/GPU acceleration enables pore-scale simulation of large systems. The pipeline developed in this thesis is fully open-source and versatile, as demonstrated by its application to different kinds of porous media, and we anticipate its future application to other reaction-diffusion problems in porous media, in particular from biology.
2023
Abhinav Singh, Ivo F. Sbalzarini, Anas Obeidat Entropically damped artificial compressibility for the discretization corrected particle strength exchange method in incompressible fluid mechanics. Computers & Fluids, 267 Art. No. 106074 (2023)
PDF
DOI
We present a consistent mesh-free numerical scheme for solving the incompressible Navier–Stokes equations. Our method is based on entropically damped artificial compressibility for imposing the incompressibility constraint explicitly, and the Discretization-Corrected Particle Strength Exchange (DC-PSE) method to consistently discretize the differential operators on mesh-free particles. We further couple our scheme with Brinkman penalization to solve the Navier–Stokes equations in complex geometries. The method is validated using the 3D Taylor–Green vortex flow and the lid-driven cavity flow problem in 2D and 3D, where we also compare our method with hr-SPH and report better accuracy for DC-PSE. In order to validate DC-PSE Brinkman penalization, we study flow past obstacles, such as a cylinder, and report excellent agreement with previous studies.
Abhinav Singh Efficient and Scalable Simulations of Active Hydrodynamics in Three Dimensions.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2023)
Open Access
Active matter represents a unique class of non-equilibrium systems, including examples ranging from cellular structures to large-scale biological tissues. These systems exhibit intriguing spatiotemporal dynamics, driven by the constituent particles’ continuous energy expenditure. Such active-matter systems, featuring complex hydrodynamics, are described by sophisticated mathematical models, typically using partial differential equations (PDEs). PDEs modeling hydrodynamics, such as the Navier-Stokes equations, are analytically intractable, and notoriously challenging to study computationally. The challenges include the need for consistent numerical methods along with their efficient and scalable high-performance computer implementation to solve the PDEs numerically. However, when considering new theoretical PDE models, such as active hydrodynamics, conventional approaches often fall short due to the specialization made in the numerical methods to study certain specific models. The inherent complexity and nonlinearity of active-matter PDEs add to the challenge. Hence, the computational study of such active-matter PDE models requires rapidly evolving high-performance computer software that can easily implement new numerical methods to solve these equations in biologically realistic three-dimensional domains. This presents a rich, yet underexplored territory demanding scalable computational frameworks that apply to a large class of PDEs. In this thesis, we introduce a computational framework that effectively allows for using multiple numerical methods through a context-aware template expression system akin to an embedded domain-specific language. This framework primarily aims at solving lengthy PDEs associated with active hydrodynamics in complex domains, while experimenting with new numerical methods. Existing PDE-solving codes often lack this flexibility, as they are closely tied to a PDE and domain geometry that rely on a specific numerical method. We overcome these limitations by using an object-oriented implementation design, and show experiments with adaptive and numerically consistent particle-based approach called Discretization-Corrected Particle Strength Exchange (DC-PSE). DC-PSE allows for the higher-order discretization of differential operators on arbitrary particle distributions leading to the possibility of solving active hydrodynamic PDEs in complex domains. However, the curse of dimensionality makes it difficult to numerically solve three-dimensional equations on single-core architectures and warrants the use of parallel and distributed computers. We design a novel template-expression system and implement it in the scalable scientific computing library OpenFPM. Our methodology offers an expression-based embedded language, enabling PDE codes to be written in a form that closely mirrors mathematical notation. Leveraging OpenFPM, this approach also ensures parallel scalability. To further enhance our framework's versatility, we employ a \textit{separation-of-concerns} abstraction, segregating the model equations from numerics, and domain geometry. This allows for the rapid rewriting of codes for agile numerical experiments across different model equations in various geometries. Supplementing this framework, we develop a distributed algebra system compatible with OpenFPM and Boost Odeint. This algebra system opens avenues for a multitude of explicit adaptive time-integration schemes, which can be selected by modifying a single line of code while maintaining parallel scalability. Motivated by symmetry-preserving theories of active hydrodynamics, and as a first benchmark of our template-expression system, we present a high-order numerically convergent scheme to study active polar fluids in arbitrary three-dimensional domains. We derive analytical solutions in simple Cartesian geometries and use them to show the numerical convergence of our algorithm. Further, we showcase the scalability of the computer code written using our expression system on distributed computing systems. To cater to the need for solving PDEs on curved surfaces, we present a novel meshfree numerical scheme, the Surface DC-PSE method. Upon implementation in our scalable framework, we benchmark Surface DC-PSE for both explicit and implicit Laplace-Beltrami operators and show applications to computing mean and Gauss curvature. Finally, we apply our computational framework to exploring the three-dimensional active hydrodynamics of biological flowing matter, a prominent model system to study the active dynamics of cytoskeletal networks, celluar migration, and tissue mechanics. Our software framework effectively tackles the challenges associated to numerically solving such non-equilibrium spatiotemporal PDEs. We perform linear perturbation analysis of the three-dimensional Ericksen-Leslie model and find an analytical expression for the critical active potential or, equivalently, a critical length of the system above which a spontaneous flow transition occurs. This spontaneous flow transition is a first realization of a three-dimensional active Fr\'eedericksz transition. With our expression system, we successfully simulate 3D active fluids, finding phases of spontaneous flow transitions, traveling waves, and spatiotemporal chaos with increasing active stress. We numerically find a topological phase transition similar to the Berezinskii–Kosterlitz–Thouless transition (BKT transition) of the two-dimensional XY model that occurs in active polar fluids after the spontaneous flow transition. We then proceed to non-Cartesian geometries and show the application of our software framework to solve the active polar fluid equations in spherical domains. We find spontaneous flows in agreement with recent experimental observations. We further showcase the framework to solve the equations in 3D annular domains and a `peanut' geometry that resembles a dividing cell. Our simulations further recapitulate the actin flows observed in \textit{Xenopus} egg extracts within spherical shell geometries, showcasing our framework's versatility in handling complex geometrical modifications of model equations. Looking ahead, we hope our framework will serve as a foundation for further advancements in computational morphogenesis, fostering collaboration and using the present techniques in biophysical modeling.
Tina Subic Gaussian Reaction Diffusion Master Equation : A Reaction Diffusion Master Equation With an Efficient Diffusion Model for Fast Exact Stochastic Simulations.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2023)
Open Access
Complex spatial structures in biology arise from random interactions of molecules. These molecular interactions can be studied using spatial stochastic models, such as Reaction Diffusion Master Equation (RDME), a mesoscopic model that subdivides the spatial domain into smaller, well mixed grid cells, in which the macroscopic diffusion-controlled reactions take place. While RDME has been widely used to study how fluctuations in number of molecules affect spatial patterns, simulations are computationally expensive and it requires a lower bound for grid cell size to avoid an apparent unphysical loss of bimolecular reactions. In this thesis, we propose Gaussian Reaction Diffusion Master Equation (GRDME), a novel model in the RDME framework, based on the discretization of the Laplace operator with Particle Strength Exchange (PSE) method with a Gaussian kernel. We show that GRDME is a computationally efficient model compared to RDME. We further resolve the controversy regarding the loss of bimolecular reactions and argue that GRDME can flexibly bridge the diffusion-controlled and ballistic regimes in mesoscopic simulations involving multiple species. To efficiently simulate GRDME, we develop Gaussian Next Subvolume Method (GNSM). GRDME simulated with GNSM up to six-times lower computational cost for a three-dimensional simulation, providing a significant computational advantage for modeling three-dimensional systems. The computational cost can be further lowered by increasing the so-called smoothing length of the Gassian jumps. We develop a guideline to estimate the grid resolution below which RDME and GRDME exhibit loss of bimolecular reactions. This loss of reactions has been considered unphysical by others. Here we show that this loss of bimolecular reactions is consistent with the well-established theory on diffusion-controlled reaction rates by Collins and Kimball, provided that the rate of bimolecular propensity is interpreted as the rate of the ballistic step, rather than the macroscopic reaction rate. We show that the reaction radius is set by the grid resolution. Unlike RDME, GRDME enables us to explicitly model various sizes of the molecules. Using this insight, we explore the diffusion-limited regime of reaction dynamics and discover that diffusion-controlled systems resemble small, discrete systems. Others have shown that a reaction system can have discreteness-induced state inversion, a phenomenon where the order of the concentrations differs when the system size is small. We show that the same reaction system also has diffusion-controlled state inversion, where the order of concentrations changes, when the diffusion is slow. In summary, we show that GRDME is a computationally efficient model, which enables us to include the information of the molecular sizes into the model.
Suryanarayana Maddu, Dominik Sturm, Bevan Cheeseman, Christian L. Müller, Ivo F. Sbalzarini STENCIL-NET for equation-free forecasting from data. Sci Rep, 13(1) Art. No. 12787 (2023)
Open AccessPDF
DOI
We present an artificial neural network architecture, termed STENCIL-NET, for equation-free forecasting of spatiotemporal dynamics from data. STENCIL-NET works by learning a discrete propagator that is able to reproduce the spatiotemporal dynamics of the training data. This data-driven propagator can then be used to forecast or extrapolate dynamics without needing to know a governing equation. STENCIL-NET does not learn a governing equation, nor an approximation to the data themselves. It instead learns a discrete propagator that reproduces the data. It therefore generalizes well to different dynamics and different grid resolutions. By analogy with classic numerical methods, we show that the discrete forecasting operators learned by STENCIL-NET are numerically stable and accurate for data represented on regular Cartesian grids. A once-trained STENCIL-NET model can be used for equation-free forecasting on larger spatial domains and for longer times than it was trained for, as an autonomous predictor of chaotic dynamics, as a coarse-graining method, and as a data-adaptive de-noising method, as we illustrate in numerical experiments. In all tests, STENCIL-NET generalizes better and is computationally more efficient, both in training and inference, than neural network architectures based on local (CNN) or global (FNO) nonlinear convolutions.
Justina Stark, Ivo F. Sbalzarini An open-source pipeline for solving continuous reaction–diffusion models in image-based geometries of porous media. J Comput Sci, 72 Art. No. 102118 (2023)
Open AccessPDF
DOI
We present a versatile open-source pipeline for simulating inhomogeneous reaction–diffusion processes in highly resolved, image-based geometries of porous media with reactive boundaries. Resolving realistic pore-scale geometries in numerical models is challenging and computationally demanding, as the scale differences between the sizes of the interstitia and the whole system can lead to prohibitive memory requirements. The present pipeline combines a level-set method with geometry-adapted sparse block grids on GPUs to efficiently simulate reaction–diffusion processes in image-based geometries. We showcase the method by applying it to fertilizer diffusion in soil, heat transfer in porous ceramics, and determining effective diffusion coefficients and tortuosity. The present approach enables solving reaction–diffusion partial differential equations in real-world geometries applicable to porous media across fields such as engineering, environmental science, and biology.
Abhinav Singh#, Alejandra Maria Foggia, Pietro Incardona, Ivo F. Sbalzarini# A Meshfree Collocation Scheme for Surface Differential Operators on Point Clouds. J Sci Comput, 96 Art. No. 89 (2023)
Open AccessPDF
DOI
We present a meshfree collocation scheme to discretize intrinsic surface differential operators over scalar fields on smooth curved surfaces with given normal vectors and a non-intersecting tubular neighborhood. The method is based on discretization-corrected particle strength exchange (DC-PSE), which generalizes finite difference methods to meshfree point clouds. The proposed Surface DC-PSE method is derived from an embedding theorem, but we analytically reduce the operator kernels along surface normals to obtain a purely intrinsic computational scheme over surface point clouds. We benchmark Surface DC-PSE by discretizing the Laplace–Beltrami operator on a circle and a sphere, and we present convergence results for both explicit and implicit solvers. We then showcase the algorithm on the problem of computing Gauss and mean curvature of an ellipsoid and of the Stanford Bunny by approximating the intrinsic divergence of the normal vector field. Finally, we compare Surface DC-PSE with surface finite elements (SFEM) and diffuse-interface finite elements (DI FEM) in a validation case.
Sarah Perez, Suryanarayana Maddu, Ivo F. Sbalzarini, Philippe Poncet Adaptive weighting of Bayesian physics informed neural networks for multitask and multiscale forward and inverse problems. J Comput Phys, 491 Art. No. 112342 (2023)
Open AccessPDF
DOI
In this paper, we present a novel methodology for automatic adaptive weighting of Bayesian Physics-Informed Neural Networks (BPINNs), and we demonstrate that this makes it possible to robustly address multi-objective and multiscale problems. BPINNs are a popular framework for data assimilation, combining the constraints of Uncertainty Quantification (UQ) and Partial Differential Equation (PDE). The relative weights of the BPINN target distribution terms are directly related to the inherent uncertainty in the respective learning tasks. Yet, they are usually manually set a-priori, that can lead to pathological behavior, stability concerns, and to conflicts between tasks which are obstacles that have deterred the use of BPINNs for inverse problems with multiscale dynamics.
The present weighting strategy automatically tunes the weights by considering the multitask nature of target posterior distribution. We show that this remedies the failure modes of BPINNs and provides efficient exploration of the optimal Pareto front. This leads to better convergence and stability of BPINN training while reducing sampling bias. The determined weights moreover carry information about task uncertainties, reflecting noise levels in the data and adequacy of the PDE model.
We demonstrate this in numerical experiments in Sobolev training, and compare them to analytically ε-optimal baseline, and in a multiscale Lotka-Volterra inverse problem. We eventually apply this framework to an inpainting task and an inverse problem, involving latent field recovery for incompressible flow in complex geometries.
Pietro Incardona, Aryaman Gupta, Serhii Yaskovets, Ivo F. Sbalzarini A portable C++ library for memory and compute abstraction on multi-core CPUs and GPUs. Concurrency Computat. Pract. Exper., 35(25) Art. No. e7870 (2023)
Open AccessPDF
DOI
We present a C++ library for transparent memory and compute abstraction across CPU and GPU architectures. Our library combines generic data structures like vectors, multi-dimensional arrays, maps, graphs, and sparse grids with basic generic algorithms like arbitrary-dimensional convolutions, copying, merging, sorting, prefix sum, reductions, neighbor search, and filtering. The memory layout of the data structures is adapted at compile time using C++ tuples with optional memory double-mapping between host and device and the capability of using memory managed by external libraries with no data copying. We combine this transparent memory layout with generic thread-parallel algorithms under two alternative common interfaces: a CUDA-like kernel interface and a lambda-function interface. We quantify the memory and compute performance and portability of our implementation using micro-benchmarks, showing that the abstractions introduce negligible performance overhead, and we compare performance against the current state of the art in a real-world scientific application from computational fluid mechanics.
Aryaman Gupta Interactive in situ visualization of large volume data.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2023)
Open Access
Three-dimensional volume data is routinely produced, at increasingly high spatial resolution, in computer simulations and image acquisition tasks. In-situ visualization, the visualization of an experiment or simulation while it is running, enables new modes of interaction, including simulation steering and experiment control. These can provide the scientist a deeper understanding of the underlying phenomena, but require interactive visualization with smooth viewpoint changes and zooming to convey depth perception and spatial understanding. As the size of the volume data increases, however, it is increasingly challenging to achieve interactive visualization with smooth viewpoint changes. This thesis presents an end-to-end solution for interactive in-situ visualization based on novel extensions proposed to the Volumetric Depth Image (VDI) representation. VDIs are view-dependent, compact representations of volume data than can be rendered faster than the original data. Novel methods are proposed in this thesis for generating VDIs on large data and for rendering them faster. Together, they enable interactive in situ visualization with smooth viewpoint changes and zooming for large volume data. The generation of VDIs involves decomposing the volume rendering integral along rays into segments that store composited color and opacity, forming a representation much smaller than the volume data. This thesis introduces a technique to automatically determine the sensitivity parameter that governs the decomposition of rays, eliminating the need for manual parameter tuning in the generation of a VDI. Further, a method is proposed for sort-last parallel generation and compositing of VDIs on distributed computers, enabling their in situ generation with distributed numerical simulations. A low latency architecture is proposed for the sharing of data and hardware resources with a running simulation. The resulting VDI can be streamed for interactive visualization. A novel raycasting method is proposed for rendering VDIs. Properties of perspective projection are exploited to simplify the intersection of rays with the view-dependent segments contained within the VDI. Spatial smoothness in volume data is leveraged to minimize memory accesses. Benchmarks are performed showing that the method significantly outperforms existing methods for rendering the VDI, and achieves responsive frame rates for High Definition (HD) display resolutions near the viewpoint of generation. Further, a method is proposed to subsample the VDI for preview rendering, maintaining high frame rates even for large viewpoint deviations. The quality and performance of the approach are analyzed on multiple datasets, and the contributions are provided as extensions of established open-source tools. The thesis concludes with a discussion on the strengths, limitations, and future directions for the proposed approach.
Abhinav Singh, Quentin Vagne, Frank Jülicher, Ivo F. Sbalzarini Spontaneous flow instabilities of active polar fluids in three dimensions. Phys Rev Research, 5 Art. No. L022061 (2023)
Open AccessPDF
DOI
Active polar fluids exhibit spontaneous flow when sufficient active stress is generated by internal molecular mechanisms. This is also referred to as an active Fréedericksz transition. Experiments have revealed the existence of competing in-plane and out-of-plane instabilities in three-dimensional active matter. So far, however, a theoretical model reconciling all observations is missing. In particular, the role of boundary conditions in these instabilities still needs to be explained. Here, we characterize the spontaneous flow transition in a symmetry-preserving three-dimensional active Ericksen-Leslie model, showing that the boundary conditions select the emergent behavior. Using nonlinear numerical solutions and linear perturbation analysis, we explain the mechanism for both in-plane and out-of-plane instabilities under extensile active stress for perpendicular polarity anchoring at the boundary, whereas parallel anchoring only permits in-plane flows under contractile stress or out-of-plane wrinkling under extensile stress.
Aryaman Gupta, Pietro Incardona, Anton Brock, Guido Reina, Steffen Frey, Stefan Gumhold, Ulrik Günther, Ivo F. Sbalzarini Parallel Compositing of Volumetric Depth Images for Interactive Visualization of Distributed Volumes at High Frame Rates.
In: Eurographics Symposium on Parallel Graphics and Visualization (EGPGV) Proceedings
(2023), The Eurographics Association (2023), 25-35
Open AccessPDF
DOI
We present a parallel compositing algorithm for Volumetric Depth Images (VDIs) of large three-dimensional volume data. Large distributed volume data are routinely produced in both numerical simulations and experiments, yet it remains challenging to visualize them at smooth, interactive frame rates. VDIs are view-dependent piecewise constant representations of volume data that offer a potential solution. They are more compact and less expensive to render than the original data. So far, however, there is no method for generating VDIs from distributed data. We propose an algorithm that enables this by sort-last parallel generation and compositing of VDIs with automatically chosen content-adaptive parameters. The resulting composited VDI can then be streamed for remote display, providing responsive visualization of large, distributed volume data.
Mateusz Susik, Ivo F. Sbalzarini Variational inference accelerates accurate DNA mixture deconvolution. Forensic Sci Int Genet, 65 Art. No. 102890 (2023)
Open AccessPDF
DOI
We investigate a class of DNA mixture deconvolution algorithms based on variational inference, and we show that this can significantly reduce computational runtimes with little or no effect on the accuracy and precision of the result. In particular, we consider Stein Variational Gradient Descent (SVGD) and Variational Inference (VI) with an evidence lower-bound objective. Both provide alternatives to the commonly used Markov-Chain Monte-Carlo methods for estimating the model posterior in Bayesian probabilistic genotyping. We demonstrate that both SVGD and VI significantly reduce computational costs over the current state of the art. Importantly, VI does so without sacrificing precision or accuracy, presenting an overall improvement over previously published methods.
Johannes Pahlke A unifying mathematical definition enables the theoretical study of the algorithmic class of particle methods.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2023)
Open Access
Mathematical definitions provide a precise, unambiguous way to formulate concepts. They also provide a common language between disciplines. Thus, they are the basis for a well-founded scientific discussion. In addition, mathematical definitions allow for deeper insights into the defined subject based on mathematical theorems that are incontrovertible under the given definition. Besides their value in mathematics, mathematical definitions are indispensable in other sciences like physics, chemistry, and computer science. In computer science, they help to derive the expected behavior of a computer program and provide guidance for the design and testing of software. Therefore, mathematical definitions can be used to design and implement advanced algorithms. One class of widely used algorithms in computer science is the class of particle-based algorithms, also known as particle methods. Particle methods can solve complex problems in various fields, such as fluid dynamics, plasma physics, or granular flows, using diverse simulation methods, including Discrete Element Methods (DEM), Molecular Dynamics (MD), Reproducing Kernel Particle Methods (RKPM), Particle Strength Exchange (PSE), and Smoothed Particle Hydrodynamics (SPH). Despite the increasing use of particle methods driven by improved computing performance, the relation between these algorithms remains formally unclear. In particular, particle methods lack a unifying mathematical definition and precisely defined terminology. This prevents the determination of whether an algorithm belongs to the class and what distinguishes the class. Here we present a rigorous mathematical definition for determining particle methods and demonstrate its importance by applying it to several canonical algorithms and those not previously recognized as particle methods. Furthermore, we base proofs of theorems about parallelizability and computational power on it and use it to develop scientific computing software. Our definition unified, for the first time, the so far loosely connected notion of particle methods. Thus, it marks the necessary starting point for a broad range of joint formal investigations and applications across fields.
Pietro Incardona, Aryaman Gupta, Serhii Yaskovets, Ivo F. Sbalzarini A C++ Library for Memory Layout and Performance Portability of Scientific Applications
In: Euro-Par 2022: Parallel Processing Workshops : Euro-Par 2022 International Workshops, Glasgow, UK, August 22–26, 2022, Revised Selected Papers
(2023) (Lecture Notes in Computer Science ; 13835), New York, Springer (2023), 109-120
DOI
We present a C++14 library for performance portability of scientific computing codes across CPU and GPU architectures. Our library combines generic data structures like vectors, multi-dimensional arrays, maps, graphs, and sparse grids with basic, reusable algorithms like convolutions, sorting, prefix sum, reductions, and scan. The memory layout of the data structures is adapted at compile-time using tuples with optional memory mirroring between CPU and GPU. We combine this transparent memory mapping with generic algorithms under two alternative programming interfaces: a CUDA-like kernel interface for multi-core CPUs, Nvidia GPUs, and AMD GPUs, as well as a lambda interface. We validate and benchmark the presented library using micro-benchmarks, showing that the abstractions introduce negligible performance overhead, and we compare performance against the current state of the art.
Mateusz Susik, Ivo F. Sbalzarini Analysis of the Hamiltonian Monte Carlo genotyping algorithm on PROVEDIt mixtures including a novel precision benchmark. Forensic Sci Int Genet, 64 Art. No. 102840 (2023)
Open AccessPDF
DOI
We provide an internal validation study of a recently published precise DNA mixture algorithm based on Hamiltonian Monte Carlo sampling (Susik et al., 2022). We provide results for all 428 mixtures analysed by Riman et al. (2021) and compare the results with two state-of-the-art software products: STRmix™ v2.6 and Euroformix v3.4.0. The comparison shows that the Hamiltonian Monte Carlo method provides reliable values of likelihood ratios (LRs) close to the other methods. We further propose a novel large-scale precision benchmark and quantify the precision of the Hamiltonian Monte Carlo method, indicating its improvements over existing solutions. Finally, we analyse the influence of the factors discussed by Buckleton et al. (2022).
Aryaman Gupta, Ulrik Günther, Pietro Incardona, Guido Reina, Steffen Frey, Stefan Gumhold, Ivo F. Sbalzarini Efficient Raycasting of Volumetric Depth Images for Remote Visualization of Large Volumes at High Frame Rates.
In: 2023 IEEE 16TH PACIFIC VISUALIZATION SYMPOSIUM, PACIFICVIS
(2023) (IEEE Pacific Visualization Symposium), Piscataway, N.J., IEEE (2023), 61-70
PDF
DOI
We present an efficient raycasting algorithm for rendering Volumetric Depth Images (VDIs), and we show how it can be used in a remote visualization setting with VDIs generated and streamed from a remote server. VDIs are compact view-dependent volume representations that enable interactive visualization of large volumes at high frame rates by decoupling viewpoint changes from expensive rendering calculations. However, current rendering approaches for VDIs struggle with achieving interactive frame rates at high image resolutions. Here, we exploit the properties of perspective projection to simplify intersections of rays with the view-dependent frustums in a VDI and leverage spatial smoothness in the volume data to minimize memory accesses. Benchmarks show that responsive frame rates can be achieved close to the viewpoint of generation for HD display resolutions, providing high-fidelity approximate renderings of Gigabyte-sized volumes. We also propose a method to subsample the VDI for preview rendering, maintaining high frame rates even for large viewpoint deviations. We provide our implementation as an extension of an established open-source visualization library.
Johannes Pahlke, Ivo F. Sbalzarini A Unifying Mathematical Definition of Particle Methods. IEEE Open J. Comput. Soc., 4 197-108 (2023)
Open AccessPDF
DOI
Particle methods are a widely used class of algorithms for computer simulation of complex phenomena in various fields, such as fluid dynamics, plasma physics, molecular chemistry, and granular flows, using diverse simulation methods, including Smoothed Particle Hydrodynamics (SPH), Particle-in-Cell (PIC) methods, Molecular Dynamics (MD), and Discrete Element Methods (DEM). Despite the increasing use of particle methods driven by improved computing performance, the relation between these algorithms remains formally unclear, and a unifying formal definition of particle methods is lacking. Here, we present a rigorous mathematical definition of particle methods and demonstrate its importance by applying it to various canonical and non-canonical algorithms, using it to prove a theorem about multi-core parallelizability, and designing a principled scientific computing software based on it. We anticipate that our formal definition will facilitate the solution of complex computational problems and the implementation of understandable and maintainable software frameworks for computer simulation.
Alicia Daeden, Alexander Mietke, Emmanuel Derivery, Carole Seum, Frank Jülicher, Marcos Gonzalez-Gaitan Polarized branched Actin modulates cortical mechanics to produce unequal-size daughters during asymmetric division. Nat Cell Biol, 25(2) 235-245 (2023)
Open Access DOI
The control of cell shape during cytokinesis requires a precise regulation of mechanical properties of the cell cortex. Only few studies have addressed the mechanisms underlying the robust production of unequal-sized daughters during asymmetric cell division. Here we report that unequal daughter-cell sizes resulting from asymmetric sensory organ precursor divisions in Drosophila are controlled by the relative amount of cortical branched Actin between the two cell poles. We demonstrate this by mistargeting the machinery for branched Actin dynamics using nanobodies and optogenetics. We can thereby engineer the cell shape with temporal precision and thus the daughter-cell size at different stages of cytokinesis. Most strikingly, inverting cortical Actin asymmetry causes an inversion of daughter-cell sizes. Our findings uncover the physical mechanism by which the sensory organ precursor mother cell controls relative daughter-cell size: polarized cortical Actin modulates the cortical bending rigidity to set the cell surface curvature, stabilize the division and ultimately lead to unequal daughter-cell size.
Sachin K. T. Veettil, Gentian Zavalani, Uwe Hernandez Acosta, Ivo F. Sbalzarini, Michael Hecht Global Polynomial Level Sets for Numerical Differential Geometry of Smooth Closed Surfaces. SIAM J Sci Comput, 45(4) Art. No. A1995-A2018 (2023)
Open Access DOI
We present a computational scheme that derives a global polynomial level set parameterization for smooth closed surfaces from a regular surface-point set and prove its uniqueness. This enables us to approximate a broad class of smooth surfaces by affine algebraic varieties. From such a global polynomial level set parameterization, differential-geometric quantities like mean and Gauss curvature can be efficiently and accurately computed. Even fourth-order terms such as the Laplacian of mean curvature are approximated with high precision. The accuracy performance results in a gain of computational efficiency, significantly reducing the number of surface points required compared to classic alternatives that rely on surface meshes or embedding grids. We mathematically derive and empirically demonstrate the strengths and the limitations of the present approach, suggesting it to be applicable to a large number of computational tasks in numerical differential geometry.
Abhinav Singh*, Philipp Suhrcke*, Pietro Incardona, Ivo F. Sbalzarini A numerical solver for active hydrodynamics in three dimensions and its application to active turbulence. Phys. Fluids, 35(10) Art. No. 105155 (2023)
Open AccessPDF
DOI
We present a higher-order convergent numerical solver for active polar hydrodynamics in three-dimensional domains of arbitrary shape, along with a scalable open-source software implementation for shared- and distributed-memory parallel computers. This enables the computational study of the nonlinear dynamics of out-of-equilibrium materials from first principles. We numerically solve the nonlinear active Ericksen–Leslie hydrodynamic equations of three-dimensional (3D) active nematics using both a meshfree and a hybrid particle-mesh method in either the Eulerian or Lagrangian frame of reference. The solver is validated against a newly derived analytical solution in 3D and implemented using the OpenFPM software library for scalable scientific computing. We then apply the presented method to studying the transition of 3D active polar fluids to spatiotemporal chaos, the emergence of coherent angular motion in a 3D annulus, and chiral vortices in symmetric and asymmetric 3D shapes resembling dividing cells. Overall, this provides a robust and efficient open-source simulation framework for 3D active matter with verified numerical convergence and scalability on parallel computers.
2022
Karl Hoffmann Robust identification of topological defects in discrete vector fields with applications to biological image data.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2022)
Open Access
Topological defects are distinguished objects in vector fields that occur in a wide range of applications, ranging from material sciences to cosmology to bio-medical imaging and fingerprint recognition. This thesis considers topological point defects, also known as singular points, of two-dimensional vector fields. Besides Euclidean vectors as representation of modulus and direction, this also includes nematic vectors that equally have a modulus but direction is replaced with a head-to-tail symmetric orientation. In both case, a singular point or topological defect is an isolated discontinuity in an otherwise continuous vector field. It is characterized by its index or topological charge, which attains integer values for polar and half-integer values for nematic vector fields. There are different yet equivalent approaches to define the index. They either base on homology groups and the Brouwer degree, or on the first fundamental group and the mapping degree, or relatedly on lifting of a loop path enclosing the singular point. The definition by lift used here translates changes in the vector field along a path into a summed change in orientation angle. This translates to topological defects in discretized vector fields, where topological charge is calculated as sum of finite angle differences along a loop path between discretization points. On closer inspection, this calculation is an estimation, and is guaranteed to yield the correct estimate only with additional assumptions, for example when the underlying continuous-domain vector field is smooth and sampled at sufficiently high spatial resolution. Otherwise, arbitrary locations and charges of topological defects are possible, which yield exactly the same discretized vectors by the periodicity of representative orientation angles. Besides, the estimated topological charge depends discontinuously on each of the discrete input vectors and exhibits discrete jumps. As application data typically is subject to noise and uncertainty, this raises the question how reliable are topological defects identified in it. The present thesis quantifies, how large perturbations of a vector field are admissible without alteration of topological defects and charges. To that end, it introduces a robustness measure for each edge in a discretization grid that are combined along loop paths. Replacing critical edges of minimal robustness within a loop path by other path segments around a minimally larger area allows targeted increase of robustness. This data-dependent method called expansion over the critical edge is iterated until a user-set robustness is satisfied. The final areas of this algorithm are shown to have minimal size and therefore maximal spatial resolution, which also adapts to the local quality of data. The areas are also given as the faces in the graph of sufficiently robust edges after deleting all vertices of degree 1 (leaves) and all their connected edges. The minimal robust areas turn out to be nested by inclusion according to their robustness threshold. This allows to tradeoff detection robustness of topological charges versus their localization accuracy, both within a selection of pre-defined loop path shapes, and for free data-dependent expansion over the critical edge. Differently from defect identification by pattern matching, there is no restriction on the charge detectable. Besides, the robustness is shown to detect the size of unordered cores of defects. Robust defect areas indicate possible defect dynamics comprising motion, defect pair generation and annihilation already from single time point data. The robustness is also applicable to irregular discretization grids thanks to its graph theoretic characterization, and an extension to curved surfaces is foreseeable. The robust data-dependent defect identification is exemplified on microscopy images of the fruit fly Drosophila melanogaster. During Dorsal Closure, a developmental process, a cell sheet called amnioserosa contracts in highly regulated manner, whereby forces are actively generated and propagated along filamentous proteins like actin. Thereby, activity level and visco-elastic properties of the tissue are linked to the topological defects in the actin orientation field. Robust detection of these reveals that the sum over robust charges is clearly positive in the hundreds, whereas the overall sum of charge without robustness consideration fluctuates around zero. Numerous charges are observed, but $\pm 1/2$ dominate and confirm the amnioserosa as nematic material despite polar molecular constituents like actin. The sizes of robust defects span three orders of magnitude, and the largest defects follow the shapes of biological cells. The size distribution decays by a power law with the power for positive defects being more negative. Time courses show slightly higher speed of motion for +1/2 defects than for -1/2 defects, an order of magnitude above material flow velocity. Experiments with a genetic modification in the protein Crumbs had shown excess contraction of the amnioserosa cell layer during development. Comparing defect velocity of these embryos to wildtype suggests that viscosity and rotational viscosity increase stronger than activity level. This hypothesis remains to be tested in a combination of experiments and simulations, yet it would not have been generated in the first place without consideration of robust defects. More generally, the presented robustness measure and optimal data-dependent identification of topological defects could benefit the analysis of defects in discretized vector fields in a variety of disciplines. The optimal data-dependent identification allows for example to calculate error distributions for charge and localization of defects. The size, shape, and nested inclusion of robust defects constitute new observables, that generate numerous follow-up questions already for the fruit fly and enable novel analyses.
Karen Soans, Ana P. Ramos, Jaydeep Sidhaye, Abhijeet Krishna, Anastasia Solomatina, Karl Hoffmann, Raimund Schlüßler, Jochen Guck, Ivo F. Sbalzarini, Carl D. Modes#, Caren Norden# Collective cell migration during optic cup formation features changing cell-matrix interactions linked to matrix topology. Curr Biol, 32(22) 4817-4831 (2022)
DOI
Cell migration is crucial for organismal development and shapes organisms in health and disease. Although a lot of research has revealed the role of intracellular components and extracellular signaling in driving single and collective cell migration, the influence of physical properties of the tissue and the environment on migration phenomena in vivo remains less explored. In particular, the role of the extracellular matrix (ECM), which many cells move upon, is currently unclear. To overcome this gap, we use zebrafish optic cup formation, and by combining novel transgenic lines and image analysis pipelines, we study how ECM properties influence cell migration in vivo. We show that collectively migrating rim cells actively move over an immobile extracellular matrix. These cell movements require cryptic lamellipodia that are extended in the direction of migration. Quantitative analysis of matrix properties revealed that the topology of the matrix changes along the migration path. These changes in matrix topologies are accompanied by changes in the dynamics of cell-matrix interactions. Experiments and theoretical modeling suggest that matrix porosity could be linked to efficient migration. Indeed, interfering with matrix topology by increasing its porosity results in a loss of cryptic lamellipodia, less-directed cell-matrix interactions, and overall inefficient migration. Thus, matrix topology is linked to the dynamics of cell-matrix interactions and the efficiency of directed collective rim cell migration during vertebrate optic cup morphogenesis.
Tina Subic, Ivo F. Sbalzarini A Gaussian jump process formulation of the reaction-diffusion master equation enables faster exact stochastic simulations. J Chem Phys, 157(19) Art. No. 194110 (2022)
Open AccessPDF
DOI
We propose a Gaussian jump process model on a regular Cartesian lattice for the diffusion part of the Reaction-Diffusion Master Equation (RDME). We derive the resulting Gaussian RDME (GRDME) formulation from analogy with a kernel-based discretization scheme for continuous diffusion processes and quantify the limits of its validity relative to the classic RDME. We then present an exact stochastic simulation algorithm for the GRDME, showing that the accuracies of GRDME and RDME are comparable, but exact simulations of the GRDME require only a fraction of the computational cost of exact RDME simulations. We analyze the origin of this speedup and its scaling with problem dimension. The benchmarks suggest that the GRDME is a particularly beneficial model for diffusion-dominated systems in three dimensional spaces, often occurring in systems biology and cell biology.
Saiyam B. Jain, Shao Zongru, Sachin K. T. Veettil, Michael Hecht Adversarial attacks for machine learning denoisers and how to resist them.
In: Emerging Topics in Artificial Intelligence (ETAI) 2022
(2022) Ch. 1220402(Eds.) Giovanni Volpe (Proceedings of SPIE ; 12204), Bellingham, USA, SPIE (2022)
DOI
Adversarial attacks rely on the instability phenomenon appearing in general for all inverse problems, e.g., image classification and reconstruction, independently of the computational scheme or method used to solve the problem. We mathematically prove and empirically show that machine learning denoisers (MLD) are not excluded. That is to prove the existence of adversarial attacks given by noise patterns making the MLD run into instability, i.e., the MLD increases the noise instead of decreasing it. We further demonstrate that adversarial retraining or classic filtering do not provide an exit strategy for this dilemma. Instead, we show that adversarial attacks can be inferred by polynomial regression. Removing the underlying inferred polynomial distribution from the total noise distribution delivers an efficient technique yielding robust MLDs that make consistent computer vision tasks such as image segmentation or classification more reliable.
Anastasia Solomatina, Alice Cezanne, Yannis Kalaidzidis, Marino Zerial, Ivo F. Sbalzarini Design centering enables robustness screening of pattern formation models. Bioinformatics, 38(Suppl 2) Art. No. ii134-ii140 (2022)
Open AccessPDF
DOI
Access to unprecedented amounts of quantitative biological data allows us to build and test biochemically accurate reaction-diffusion models of intracellular processes. However, any increase in model complexity increases the number of unknown parameters and, thus, the computational cost of model analysis. To efficiently characterize the behavior and robustness of models with many unknown parameters remains, therefore, a key challenge in systems biology.
Mateusz Susik, Holger Schönborn, Ivo F. Sbalzarini Hamiltonian Monte Carlo with strict convergence criteria reduces run-to-run variability in forensic DNA mixture deconvolution. Forensic Sci Int Genet, 60 Art. No. 102744 (2022)
Open AccessPDF
DOI
Analysing mixed DNA profiles is a common task in forensic genetics. Due to the complexity of the data, such analysis is often performed using Markov Chain Monte Carlo (MCMC)-based genotyping algorithms. These trade off precision against execution time. When default settings (including default chain lengths) are used, as large as a 10-fold changes in inferred log-likelihood ratios (LR) are observed when the software is run twice on the same case. So far, this uncertainty has been attributed to the stochasticity of MCMC algorithms. Since LRs translate directly to strength of the evidence in a criminal trial, forensic laboratories desire LR with small run-to-run variability.
Suryanarayana Maddu, Bevan Cheeseman, Ivo F. Sbalzarini, Christian L. Müller Stability selection enables robust learning of differential equations from limited noisy data. Proc Roy Soc A, 478(2262) Art. No. 20210916 (2022)
Open AccessPDF
DOI
We present a statistical learning framework for robust identification of differential equations from noisy spatio-temporal data. We address two issues that have so far limited the application of such methods, namely their robustness against noise and the need for manual parameter tuning, by proposing stability- based model selection to determine the level of regularization required for reproducible inference. This avoids manual parameter tuning and improves robustness against noise in the data. Our stability selection approach, termed PDE-STRIDE, can be combined with any sparsity-promoting regression method and provides an interpretable criterion for model component importance. We show that the particular combination of stability selection with the iterative hard-thresholding algorithm from compressed sensing provides a fast and robust framework for equation inference that outperforms previous approaches with respect to accuracy, amount of data required, and robustness. We illustrate the performance of PDE-STRIDE on a range of simulated benchmark problems, and we 2 demonstrate the applicability of PDE-STRIDE on real-world data by considering purely data-driven inference of the protein interaction network for embryonic polarization in Caenorhabditis elegans. Using fluorescence microscopy images of C. elegans zygotes as input data, PDE-STRIDE is able to learn the molecular interactions of the proteins.
Joel Jonsson, Bevan Cheeseman, Suryanarayana Maddu, Krzysztof Gonciarz, Ivo F. Sbalzarini Parallel Discrete Convolutions on Adaptive Particle Representations of Images. IEEE Trans Image Process, 31 4197-4212 (2022)
Open AccessPDF
DOI
We present data structures and algorithms for native implementations of discrete convolution operators over Adaptive Particle Representations (APR) of images on parallel computer architectures. The APR is a content-adaptive image representation that locally adapts the sampling resolution to the image signal. It has been developed as an alternative to pixel representations for large, sparse images as they typically occur in fluorescence microscopy. It has been shown to reduce the memory and runtime costs of storing, visualizing, and processing such images. This, however, requires that image processing natively operates on APRs, without intermediately reverting to pixels. Designing efficient and scalable APR-native image processing primitives, however, is complicated by the APR's irregular memory structure. Here, we provide the algorithmic building blocks required to efficiently and natively process APR images using a wide range of algorithms that can be formulated in terms of discrete convolutions. We show that APR convolution naturally leads to scale-adaptive algorithms that efficiently parallelize on multi-core CPU and GPU architectures. We quantify the speedups in comparison to pixel-based algorithms and convolutions on evenly sampled data. We achieve pixel-equivalent throughputs of up to 1TB/s on a single Nvidia GeForce RTX 2080 gaming GPU, requiring up to two orders of magnitude less memory than a pixel-based implementation.
Huw Colin-York#, John M Heddleston, Eric Wait, Narain Karedla, Michael deSantis, Satya Khuon, Teng-Leong Chew, Ivo F. Sbalzarini, Marco Fritzsche# Quantifying Molecular Dynamics within Complex Cellular Morphologies using LLSM-FRAP. Small Methods, 6 Art. No. 2200149 (2022)
Open AccessPDF
DOI
Quantifying molecular dynamics within the context of complex cellular morphologies is essential toward understanding the inner workings and function of cells. Fluorescence recovery after photobleaching (FRAP) is one of the most broadly applied techniques to measure the reaction diffusion dynamics of molecules in living cells. FRAP measurements typically restrict themselves
to single-plane image acquisition within a subcellular-sized region of interest due to the limited temporal resolution and undesirable photobleaching induced by 3D fluorescence confocal or widefield microscopy. Here, an experimental and computational pipeline combining lattice light sheet microscopy, FRAP, and numerical simulations, offering rapid and minimally invasive quantification of molecular dynamics with respect to 3D cell morphology is presented. Having the opportunity to accurately measure and interpret the dynamics of molecules in 3D with respect to cell morphology has the potential to reveal unprecedented insights into the function of living cells.
Suryanarayana Maddu, Dominik Sturm, Christian L. Müller, Ivo F. Sbalzarini Inverse Dirichlet weighting enables reliable training of physics informed neural networks. Mach. Learn.: Sci. Technol., 3(1) Art. No. 015026 (2022)
Open AccessPDF
DOI
We characterize and remedy a failure mode that may arise from multi-scale dynamics with scale imbalances during training of deep neural networks, such as physics informed neural networks (PINNs). PINNs are popular machine-learning templates that allow for seamless integration of physical equation models with data. Their training amounts to solving an optimization problem over a weighted sum of data-fidelity and equation-fidelity objectives. Conflicts between objectives can arise from scale imbalances, heteroscedasticity in the data, stiffness of the physical equation, or from catastrophic interference during sequential training. We explain the training pathology arising from this and propose a simple yet effective inverse Dirichlet weighting strategy to alleviate the issue. We compare with Sobolev training of neural networks, providing the baseline of analytically ε-optimal training. We demonstrate the effectiveness of inverse Dirichlet weighting in various applications, including a multi-scale model of active turbulence, where we show orders of magnitude improvement in accuracy and convergence over conventional PINN training. For inverse modeling using sequential training, we find that inverse Dirichlet weighting protects a PINN against catastrophic forgetting.
Nesrine Khouzami, Friedrich Michel, Pietro Incardona, Jeronimo Castrillon, Ivo F. Sbalzarini Model-based autotuning of discretization methods in numerical simulations of partial differential equations. J Comput Sci, 57 Art. No. 101489 (2022)
Open AccessPDF
DOI
We present an autotuning approach for compile-time optimization of numerical discretization methods insimulations of partial differential equations. Our approach is based on data-driven regression of performancemodels for numerical methods. We use these models at compile time to automatically determine the parameters(e.g., resolution, time step size, etc.) of numerical simulations of continuum spatio-temporal models in order tooptimize the tradeoff between simulation accuracy and runtime. The resulting autotuner is developed for thecompiler of a Domain-Specific Language (DSL) for numerical simulations. The abstractions in the DSL enablethe compiler to automatically determine the performance models and know which discretization parametersto tune. We demonstrate that this high-level approach can explore a large space of possible simulations, withsimulation runtimes spanning multiple orders of magnitude. We evaluate our approach in two test cases:the linear diffusion equation and the nonlinear Gray-Scott reaction–diffusion equation. The results show thatour model-based autotuner consistently finds configurations that outperform those found by state-of-the-artgeneral-purpose autotuners. Specifically, our autotuner yields simulations that are on average 4.2x faster thanthose found by the best generic exploration algorithms, while using 16x less tuning time. Compared to manualtuning by a group of researchers with varying levels of expertise, the autotuner was slower than the best usersby not more than a factor of 2, whereas it was able to significantly outperform half of them.
2021
Karl Hoffmann, Ivo F. Sbalzarini Estimation of unordered core size using a robustness measure for topological defects in discretized orientation and vector fields. Proc Appl Math Mech, 21(1) Art. No. e202100105 (2021)
Open AccessPDF
DOI
We show how the finite sizes of unordered defect cores in discretized orientation and vector fields can reliably be estimated
using a robustness measure for topological defects. Topological defects, or singular points, in vector and orientation fields
are considered in applications from material science to life sciences to fingerprint recognition. Their identification from dis-
cretized two-dimensional fields must deal with discontinuities, since the estimated topological charge jumps in (half-)integer
steps upon orientation changes above a certain threshold. We use a recently proposed robustness measure [Hoffmann &
Sbalzarini, Phys. Rev. E 103(1), 012602 (2021)] that exploits this effect to quantify the influence of noise in a vector field,
and of the path chosen for defect estimation, on the detection reliability in two-dimensional discrete domains. Here, we show
how this robustness measure can be used to quantify the sizes of unordered regions surrounding a defect, which are known
as unordered cores. We suggest that the size of an unordered core can be identified as the smallest path radius of sufficient
robustness. The resulting robust core-size estimation complements singular point and index estimation and may serve as
uncertainty quantification of defect localization, or as an additional feature for defect characterization.
Anastasia Solomatina A computational framework for multidimensional parameter space screening of reaction-diffusion models in biology.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2021)
Abhinav Singh, Pietro Incardona, Ivo F. Sbalzarini A C++ expression system for partial differential equations enables generic simulations of biological hydrodynamics. Eur Phys J E Soft Matter, 44(9) Art. No. 117 (2021)
Open AccessPDF
DOI
We present a user-friendly and intuitive C++ expression system to implement numerical simulations of continuum biological hydrodynamics. The expression system allows writing simulation programs in near-mathematical notation and makes codes more readable, more compact, and less error-prone. It also cleanly separates the implementation of the partial differential equation model from the implementation of the numerical methods used to discretize it. This allows changing either of them with minimal changes to the source code. The presented expression system is implemented in the high-performance computing platform OpenFPM, supporting simulations that transparently parallelize on multi-processor computer systems. We demonstrate that our expression system makes it easier to write scalable codes for simulating biological hydrodynamics in space and time. We showcase the present framework in numerical simulations of active polar fluids, as well as in classic simulations of fluid dynamics from the incompressible Navier-Stokes equations to Stokes flow in a ball. The presented expression system accelerates scalable simulations of spatio-temporal models that encode the physics and material properties of tissues in order to algorithmically study morphogenesis.
Suryanarayana Maddu Kondaiah Data-Driven Modelling and Simulation of Spatiotemporal Processes with a View Toward Applications in Biology.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2021)
Open Access
Mathematical modeling and simulation has emerged as a fundamental means to understand physical process around us with countless real-world applications in applied science and engineering problems. However, heavy reliance on first principles, symmetry relations, and conservation laws has limited its applicability to a few scientific domains and even few real-world scenarios. Especially in disciplines like biology the underlying living constituents exhibit a myriad of complexities like non-linearities, non-equilibrium physics, self-organization and plasticity that routinely escape mathematical treatment based on governing laws. Meanwhile, recent decades have witnessed rapid advancement in computing hardware, sensing technologies, and algorithmic innovations in machine learning. This progress has helped propel data-driven paradigms to achieve unprecedented practical success in the fields of image processing and computer vision, natural language processing, autonomous transport, and etc. In the current thesis, we explore, apply, and advance statistical and machine learning strategies that help bridge the gap between data and mathematical models, with a view toward modeling and simulation of spatiotemporal processes in biology. As first, we address the problem of learning interpretable mathematical models of biologial process from limited and noisy data. For this, we propose a statistical learning framework called PDE-STRIDE based on the theory of stability selection and ℓ0-based sparse regularization for parsimonious model selection. The PDE-STRIDE framework enables model learning with relaxed dependencies on tuning parameters, sample-size and noise-levels. We demonstrate the practical applicability of our method on real-world data by considering a purely data-driven re-evaluation of the advective triggering hypothesis explaining the embryonic patterning event in the C. elegans zygote. As a next natural step, we extend our PDE-STRIDE framework to leverage prior knowledge from physical principles to learn biologically plausible and physically consistent models rather than models that simply fit the data best. For this, we modify the PDE-STRIDE framework to handle structured sparsity constraints for grouping features which enables us to: 1) enforce conservation laws, 2) extract spatially varying non-observables, 3) encode symmetry relations associated with the underlying biological process. We show several applications from systems biology demonstrating the claim that enforcing priors dramatically enhances the robustness and consistency of the data-driven approaches. In the following part, we apply our statistical learning framework for learning mean-field deterministic equations of active matter systems directly from stochastic self-propelled active particle simulations. We investigate two examples of particle models which differs in the microscopic interaction rules being used. First, we consider a self-propelled particle model endowed with density-dependent motility character. For the chosen hydrodynamic variables, our data-driven framework learns continuum partial differential equations that are in excellent agreement with analytical derived coarse-grain equations from Boltzmann approach. In addition, our structured sparsity framework is able to decode the hidden dependency between particle speed and the local density intrinsic to the self-propelled particle model. As a second example, the learning framework is applied for coarse-graining a popular stochastic particle model employed for studying the collective cell motion in epithelial sheets. The PDE-STRIDE framework is able to infer novel PDE model that quantitatively captures the flow statistics of the particle model in the regime of low density fluctuations. Modern microscopy techniques produce GigaBytes (GB) and TeraBytes (TB) of data while imaging spatiotemporal developmental dynamics of living organisms. However, classical statistical learning based on penalized linear regression models struggle with issues like accurate computation of derivatives in the candidate library and problems with computational scalability for application to “big” and noisy data-sets. For this reason we exploit the rich parameterization of neural networks that can efficiently learn from large data-sets. Specifically, we explore the framework of Physics-Informed Neural Networks (PINN) that allow for seamless integration of physics priors with measurement data. We propose novel strategies for multi-objective optimization that allow for adapting PINN architecture to multi-scale modeling problems arising in biology. We showcase application examples for both forward and inverse modeling of mesoscale active turbulence phenomenon observed in dense bacterial suspensions. Employing our strategies, we demonstrate orders of magnitude gain in accuracy and convergence in comparison with conventional formulation for solving multi-objective optimization in PINNs. In the concluding chapter of the thesis, we skip model interpretability and focus on learning computable models directly from noisy data for the purpose of pure dynamics forecasting. We propose STENCIL-NET, an artificial neural network architecture that learns solution adaptive spatial discretization of an unknown PDE model that can be stably integrated in time with negligible loss in accuracy. To support this claim, we present numerical experiments on long-term forecasting of chaotic PDE solutions on coarse spatio-temporal grids, and also showcase de-noising application that help decompose spatiotemporal dynamics from the noise in an equation-free manner.
Pietro Incardona, Tommaso Bianucci, Ivo F. Sbalzarini Distributed Sparse Block Grids on GPUs.
In: High Performance Computing : 36th International Conference, ISC High Performance 2021, Virtual Event, June 24 – July 2, 2021, Proceedings
(2021) (Lecture Notes in Computer Science ; 12728), Cham, Springer International Publishing (2021), 272-290
DOI
We present a design and implementation of distributed sparse block grids that transparently scale from a single CPU to multi-GPU clusters. We support dynamic sparse grids as, e.g., occur in computer graphics with complex deforming geometries and in multi-resolution numerical simulations. We present the data structures and algorithms of our approach, focusing on the optimizations required to render them computationally efficient on CPUs and GPUs alike. We provide a scalable implementation in the OpenFPM software library for HPC. We benchmark our implementation on up to 16 Nvidia GTX 1080 GPUs and up to 64 Nvidia A100 GPUs showing state-of-the-art scalability (68% to 96% parallel efficiency) on three benchmark problems. On a single GPU, our implementation is 14 to 140-fold faster than on a multi-core CPU.
Nesrine Khouzami, Lars Schütze, Pietro Incardona, Landfried Kraatz, Tina Subic, Jeronimo Castrillon, Ivo F. Sbalzarini The OpenPME Problem Solving Environment for Numerical Simulations.
In: Computational Science – ICCS 2021 21st International Conference, Krakow, Poland, June 16–18, 2021, Proceedings, Part I
(2021) (Lecture Notes in Computer Science ; 12742), Cham, Springer International Publishing (2021), 614-627
DOI
Michael Hecht, Ivo F. Sbalzarini Biggs Theorem for Directed Cycles and Topological Invariants of Digraphs. Adv Pure Math, 11(6) 573-594 (2021)
Open AccessPDF
DOI
We generalize Biggs Theorem to the case of directed cycles of multi-digraphs allowing to compute the dimension of the directed cycle space independently of the graph representation with linear runtime complexity. By considering two-dimensional CW complex of elementary cycles and deriving formulas for the Betti numbers of the associated cellular homology groups, we extend the list of representation independent topological inavariants measuring the graph structure. We prove the computation of the 2nd Betti number to be sharp #Phard in general and present specific representation invariant sub-fillings yielding efficiently computable homology groups. Finally, we suggest howto use the provided structural measures to shed new light on graph theoretical problems as graph embeddings, discrete Morse theoryand graph clustering.
Anastasia Solomatina, Yannis Kalaidzidis, Alice Cezanne, Karen Soans, Caren Norden, Marino Zerial, Ivo F. Sbalzarini Particle-based Segmentation of Extended Objects on Curved Biological Membranes.
In: ISBI 2021, IEEE International Symposium on Biomedical Imaging (ISBI)
(2021), Piscataway, N.J., IEEE (2021), 1150-1154
DOI
We present a novel method for model-based segmentation of extended, blob-like objects on curved surfaces. Our method addresses several challenges arising when imaging curved biological membrane, such as out-of-membrane signal and geometry-induced background variations. We use a particle-based reconstruction of the membrane geometry, moment-conserving intensity interpolation from pixels to surface particles, and model-based in-surface segmentation. Our method denoises and deconvolves images, corrects for background variations, and quantifies the number, size, and intensity of segmented objects. We benchmark the accuracy of the method and present two applications to (1) neuroepithelial focal adhesion sites during optic cup morphogenesis in zebrafish and (2) reconstituted membrane domains bearing the small GTPase Rab5 on spherical beads.
Suryanarayana Maddu, Bevan Cheeseman, Christian L. Müller, Ivo F. Sbalzarini Learning physically consistent differential equation models from data using group sparsity. Phys Rev E, 103(4) Art. No. 042310 (2021)
Open AccessPDF
DOI
We propose a statistical learning framework based on group-sparse regression that can be used to (i) enforce
conservation laws, (ii) ensure model equivalence, and (iii) guarantee symmetries when learning or inferring
differential-equation models from data. Directly learning interpretable mathematical models from data has
emerged as a valuable modeling approach. However, in areas such as biology, high noise levels, sensor-induced
correlations, and strong intersystem variability can render data-driven models nonsensical or physically inconsistent
without additional constraints on the model structure. Hence, it is important to leverage prior knowledge
from physical principles to learn biologically plausible and physically consistent models rather than models that
simply fit the data best. We present the group iterative hard thresholding algorithm and use stability selection to
infer physically consistent models with minimal parameter tuning. We show several applications from systems
biology that demonstrate the benefits of enforcing priors in data-driven modeling.
Cameron Arshadi, Ulrik Günther, Mark Eddison, Kyle Harrington, Tiago A Ferreira SNT: a unifying toolbox for quantification of neuronal anatomy. Nat Methods, 18(4) 374-377 (2021)
DOI
SNT is an end-to-end framework for neuronal morphometry and whole-brain connectomics that supports tracing, proof-editing, visualization, quantification and modeling of neuroanatomy. With an open architecture, a large user base, community-based documentation, support for complex imagery and several model organisms, SNT is a flexible resource for the broad neuroscience community. SNT is both a desktop application and multi-language scripting library, and it is available through the Fiji distribution of ImageJ.
Benjamin Dalton, Ivo F. Sbalzarini, Itsuo Hanasaki Fundamentals of the logarithmic measure for revealing multimodal diffusion. Biophys J, 120(5) 829-843 (2021)
Open AccessPDF
DOI
We develop a theoretical foundation for a time-series analysis method suitable for revealing the spectrum of diffusion coefficients in mixed Brownian systems, where no prior knowledge of particle distinction is required. This method is directly relevant for particle tracking in biological systems, where diffusion processes are often non-uniform. We transform Brownian data onto the logarithmic domain, where the coefficients for individual modes of diffusion appear as distinct spectral peaks in the probability density. We refer to the method as the logarithmic measure of diffusion, or simply as the logarithmic measure. We provide a general protocol for deriving analytical expressions for the probability densities on the logarithmic domain. The protocol is applicable for any number of spatial dimensions with any number of diffusive states. The analytical form can be fitted to data to reveal multiple diffusive modes. We validate the theoretical distributions and benchmark the accuracy and sensitivity of the method by extracting multi-modal diffusion coefficients from 2D Brownian simulations of poly-disperse filament bundles. Bundling the filaments allows us to control the system non-uniformity and hence quantify the sensitivity of the method. By exploiting the anisotropy of the simulated filaments, we generalize the logarithmic measure to rotational diffusion. By fitting the analytical forms to simulation data, we confirm the method's theoretical foundation. An error analysis in the single-mode regime shows that the proposed method is comparable in accuracy to the standard mean squared displacement approach for evaluating diffusion coefficients. For the case of multi-modal diffusion, we compare the logarithmic measure against other more sophisticated methods, showing that both model selectivity and extraction accuracy are comparable for small data sets. Therefore we suggest that the logarithmic measure, as a method for multi-modal diffusion coefficient extraction, is ideally suited for small data sets, a condition often confronted in the experimental context. Finally, we critically discuss the proposed benefits of the method and its information content.
Suryanarayana Maddu, Dominik Sturm, Bevan Cheeseman, Christian L. Müller, Ivo F. Sbalzarini Learning Computable Models from Data.
In: 14th World Congress in Computational Mechanics (WCCM), ECCOMAS Congress 2020, 11-15 January 2021, Virtual Congress
(2021), Barcelona, International Center for Numerical Methods in Engineering (CIMNE) (2021), 1-6
PDF
DOI
Michael Hecht, Krzysztof Gonciarz, Szabolcs Horvát Tight Localizations of Feedback Sets. ACM J Exp Algorithmics, 26(1) Art. No. 1.5 (2021)
Open AccessPDF
DOI
The classical NP–hard feedback arc set problem (FASP) and feedback vertex set problem (FVSP) ask for a minimum set of arcs ε ⊆ E or vertices ν ⊆ V whose removal G ∖ ε, G ∖ ν makes a given multi–digraph G=(V, E) acyclic, respectively. Though both problems are known to be APX–hard, constant ratio approximations or proofs of inapproximability are unknown. We propose a new universal O(|V||E|4)–heuristic for the directed FASP. While a ratio of r ≈ 1.3606 is known to be a lower bound for the APX–hardness, at least by empirical validation we achieve an approximation of r ≤ 2. Most of the relevant applications, such as circuit testing, ask for solving the FASP on large sparse graphs, which can be done efficiently within tight error bounds with our approach.
Karl Hoffmann, Ivo F. Sbalzarini Robustness of topological defects in discrete domains Phys Rev E, 103(1) Art. No. 012602 (2021)
Open AccessPDF
DOI
2020
Aryaman Gupta, Pietro Incardona, Ata Deniz Aydin, Stefan Gumhold, Ulrik Günther, Ivo F. Sbalzarini An Architecture for Interactive In Situ Visualization and its Transparent Implementation in OpenFPM.
In: ISAV'20 In Situ Infrastructures for Enabling Extreme-Scale Analysis and Visualization
(2020), New York, ACM (2020), 20-26
Open AccessPDF
DOI
Live in situ visualization of numerical simulations – interactive visualization while the simulation is running – can enable new modes of interaction, including computational steering. Designing easy-to-use distributed in situ architectures, with viewing latency low enough, and frame rate high enough, for interactive use, is challenging. Here, we propose a fully asynchronous, hybrid CPU–GPU in situ architecture that emphasizes interactivity. We also present a transparent implementation of this architecture embedded into the OpenFPM simulation framework. The benchmarks show that our architecture minimizes visual latencies, and achieves frame rates between 6 and 60 frames/second – depending on simulation data size and degree of parallelism – by changing only a few lines of an existing simulation code.
Ulrik Günther, Kyle Harrington, Raimund Dachselt, Ivo F. Sbalzarini Bionic Tracking: Using Eye Tracking to Track Biological Cells in Virtual Reality.
In: Computer vision - ECCV 2020 workshops : Glasgow, UK, August 23-28, 2020 : proceedings : Part 1
(2020)(Eds.) Adrien Bartoli (Lecture notes in computer science ; 12535), Cham, Springer International Publishing (2020), 280-297
PDF
DOI
We present Bionic Tracking, a novel method for solving biological cell tracking problems with eye tracking in virtual reality using commodity hardware. Using gaze data, and especially smooth pursuit eye movements, we are able to track cells in time series of 3D volumetric datasets. The problem of tracking cells is ubiquitous in developmental biology, where large volumetric microscopy datasets are acquired on a daily basis, often comprising hundreds or thousands of time points that span hours or days. The image data, however, is only a means to an end, and scientists are often interested in the reconstruction of cell trajectories and cell lineage trees. Reliably tracking cells in crowded three-dimensional space over many time points remains an open problem, and many current approaches rely on tedious manual annotation or curation. In the Bionic Tracking approach, we substitute the usual 2D point-and-click interface for annotation or curation with eye tracking in a virtual reality headset, where users follow cells with their eyes in 3D space in order to track them. We detail the interaction design of our approach and explain the graph-based algorithm used to connect different time points, also taking occlusion and user distraction into account. We demonstrate Bionic Tracking using examples from two different biological datasets. Finally, we report on a user study with seven cell tracking experts, highlighting the benefits and limitations of Bionic Tracking compared to point-and-click interfaces.
Quentin Vagne*, Jean-Patrick Vrel*, Pierre Sens A minimal self-organisation model of the Golgi apparatus. Elife, 9 Art. No. e47318 (2020)
Open Access DOI
The design principles dictating the spatio-temporal organisation of eukaryotic cells, and in particular the mechanisms controlling the self-organisation and dynamics of membrane-bound organelles such as the Golgi apparatus, remain elusive. Although this organelle was discovered 120 years ago, such basic questions as whether vesicular transport through the Golgi occurs in an anterograde (from entry to exit) or retrograde fashion are still strongly debated. Here, we address these issues by studying a quantitative model of organelle dynamics that includes: de-novo compartment generation, inter-compartment vesicular exchange, and biochemical conversion of membrane components. We show that anterograde or retrograde vesicular transports are asymptotic behaviors of a much richer dynamical system. Indeed, the structure and composition of cellular compartments and the directionality of vesicular exchange are intimately linked. They are emergent properties that can be tuned by varying the relative rates of vesicle budding, fusion and biochemical conversion.
Alice Cezanne, Janelle Lauer, Anastasia Solomatina, Ivo F. Sbalzarini, Marino Zerial A non-linear system patterns Rab5 GTPase on the membrane. Elife, 9 Art. No. e54434 (2020)
Open AccessPDF
DOI
Proteins can self-organize into spatial patterns via non-linear dynamic interactions on cellular membranes. Modelling and simulations have shown that small GTPases can generate patterns by coupling guanine nucleotide exchange factors (GEF) to effectors, generating a positive feedback of GTPase activation and membrane recruitment. Here, we reconstituted the patterning of the small GTPase Rab5 and its GEF/effector complex Rabex5/Rabaptin5 on supported lipid bilayers. We demonstrate a 'handover' of Rab5 from Rabex5 to Rabaptin5 upon nucleotide exchange. A minimal system consisting of Rab5, RabGDI and a complex of full length Rabex5/Rabaptin5 was necessary to pattern Rab5 into membrane domains. Rab5 patterning required a lipid membrane composition mimicking that of early endosomes, with PI(3)P enhancing membrane recruitment of Rab5 and acyl chain packing being critical for domain formation. The prevalence of GEF/effector coupling in nature suggests a possible universal system for small GTPase patterning involving both protein and lipid interactions.
Ulrik Günther A Modular and Open-Source Framework for Virtual Reality Visualisation and Interaction in Bioimaging.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2020)
Ivo F. Sbalzarini Systems Biology : Will Informatics also revolutionise clinical Research? - Systembiologie: Wird die Informatik auch die klinische Forschung revolutionieren? Allergologie , 43(8) 338-339 (2020)
DOI
Karl Hoffmann, Ivo F. Sbalzarini A robustness measure for singular point and index estimation in discretized orientation and vector fields. Proc Appl Math Mech, 20(1) Art. No. e202000261 (2020)
Open AccessPDF
DOI
2019
Alexander Mietke, V Jemseena, K Vijay Kumar, Ivo F. Sbalzarini, Frank Jülicher Minimal Model of Cellular Symmetry Breaking. Phys Rev Lett, 123(18) Art. No. 188101 (2019)
DOI
The cell cortex, a thin film of active material assembled below the cell membrane, plays a key role in cellular symmetry-breaking processes such as cell polarity establishment and cell division. Here, we present a minimal model of the self-organization of the cell cortex that is based on a hydrodynamic theory of curved active surfaces. Active stresses on this surface are regulated by a diffusing molecular species. We show that coupling of the active surface to a passive bulk fluid enables spontaneous polarization and the formation of a contractile ring on the surface via mechanochemical instabilities. We discuss the role of external fields in guiding such pattern formation. Our work reveals that key features of cellular symmetry breaking and cell division can emerge in a minimal model via general dynamic instabilities.
S Zitz, A Scagliarini#, Suryanarayana Maddu, A A Darhuber, J Harting# Lattice Boltzmann method for thin-liquid-film hydrodynamics. Phys Rev E, 100(3-1) Art. No. 033313 (2019)
DOI
We propose an approach to the numerical simulation of thin-film flows based on the lattice Boltzmann method. We outline the basic features of the method, show in which limits the expected thin-film equations are recovered, and perform validation tests. The numerical scheme is applied to the viscous Rayleigh-Taylor instability of a thin film and to the spreading of a sessile drop toward its equilibrium contact angle configuration. We show that the Cox-Voinov law is satisfied and that the effect of a tunable slip length on the substrate is correctly captured. We address, then, the problem of a droplet sliding on an inclined plane, finding that the Capillary number scales linearly with the Bond number, in agreement with experimental results. At last, we demonstrate the ability of the method to handle heterogenous and complex systems by showcasing the controlled dewetting of a thin film on a chemically structured substrate.
Pietro Incardona, Antonio Leo, Yaroslav Zaluzhnyi, Rajesh Ramaswamy, Ivo F. Sbalzarini OpenFPM: A scalable open framework for particle and particle-mesh codes on parallel computers Comput Phys Commun, 241 155-177 (2019)
Open AccessPDF
DOI
Ilaria Rossetti, Laura Zambusi, Paola Maccioni, Roberta Sau, Luciano Provini, M Paola Castelli, Krzysztof Gonciarz, Giancarlo Colombo, Stefano Morara Predisposition to Alcohol Drinking and Alcohol Consumption Alter Expression of Calcitonin Gene-Related Peptide, Neuropeptide Y, and Microglia in Bed Nucleus of Stria Terminalis in a Subnucleus-Specific Manner. Front Cell Neurosci, 13 Art. No. 158 (2019)
Open Access DOI
Excessive alcohol consumption is often linked to anxiety states and has a major relay center in the anterior part of bed nucleus of stria terminalis (BNST). We analyzed the impact of (i) genetic predisposition to high alcohol preference and consumption, and (ii) alcohol intake on anterior BNST, namely anterolateral (AL), anteromedial (AM), and anteroventral (lateral + medial subdivisions: AVl, AVm) subnuclei. We used two rat lines selectively bred for low- and high-alcohol preference and consumption, named Sardinian alcohol-non preferring (sNP) and -preferring (sP), respectively, the latter showing also inherent anxiety-related behaviors. We analyzed the modulation of calcitonin gene-related peptide (CGRP; exerting anxiogenic effects in BNST), neuropeptide Y (NPY; exerting mainly anxiolytic effects), and microglia activation (neuroinflammation marker, thought to increase anxiety). Calcitonin gene-related peptide-immunofluorescent fibers/terminals did not differ between alcohol-naive sP and sNP rats. Fiber/terminal NPY-immunofluorescent intensity was lower in BNST-AM and BNST-AVm of alcohol-naive sP rats. Activation of microglia (revealed by morphological analysis) was decreased in BNST-AM and increased in BNST-AVm of alcohol-naive sP rats. Prolonged (30 consecutive days), voluntary alcohol intake under the homecage 2-bottle "alcohol vs. water" regimen strongly increased CGRP intensity in BNST of sP rats in a subnucleus-specific manner: in BNST-AL, BNST-AVm, and BNST-AM. CGRP area sum, however, decreased in BNST-AM, without changes in other subnuclei. Alcohol consumption increased NPY expression, in a subnucleus-specific manner, in BNST-AL, BNST-AVl, and BNST-AVm. Alcohol consumption increased many size/shapes parameters in microglial cells, indicative of microglia de-activation. Finally, microglia density was increased in ventral anterior BNST (BNST-AVl, BNST-AVm) by alcohol consumption. In conclusion, genetic predisposition of sP rats to high alcohol intake could be in part mediated by anterior BNST subnuclei showing lower NPY expression and differential microglia activation. Alcohol intake in sP rats produced complex subnucleus-specific changes in BNST, affecting CGRP/NPY expression and microglia and leading to hypothesize that these changes might contribute to the anxiolytic effects of voluntarily consumed alcohol repeatedly observed in sP rats.
Alexander Mietke, Frank Jülicher#, Ivo F. Sbalzarini# Self-organized shape dynamics of active surfaces. Proc Natl Acad Sci U.S.A., 116(1) 29-34 (2019)
Open Access DOI
Mechanochemical processes in thin biological structures, such as the cellular cortex or epithelial sheets, play a key role during the morphogenesis of cells and tissues. In particular, they are responsible for the dynamical organization of active stresses that lead to flows and deformations of the material. Consequently, advective transport redistributes force-generating molecules and thereby contributes to a complex mechanochemical feedback loop. It has been shown in fixed geometries that this mechanism enables patterning, but the interplay of these processes with shape changes of the material remains to be explored. In this work, we study the fully self-organized shape dynamics using the theory of active fluids on deforming surfaces and develop a numerical approach to solve the corresponding force and torque balance equations. We describe the spontaneous generation of nontrivial surface shapes, shape oscillations, and directed surface flows that resemble peristaltic waves from self-organized, mechanochemical processes on the deforming surface. Our approach provides opportunities to explore the dynamics of self-organized active surfaces and can help to understand the role of shape as an integral element of the mechanochemical organization of morphogenetic processes.
Ulrik Günther, Tobias Pietzsch, Aryaman Gupta, Kyle Harrington, Pavel Tomancak, Stefan Gumhold, Ivo F. Sbalzarini scenery: Flexible Virtual Reality Visualization on the Java VM.
In: 2019 IEEE Visualization Conference (VIS)
(2019), Piscataway, N.J., IEEE (2019), 166-170
Ivo F. Sbalzarini Big-Data Analytics transformiert die Lebenswissenschaften. Informatik Spektrum, 42(6) 394-400 (2019)
Open AccessPDF
DOI
Aryaman Gupta, Ulrik Günther, Pietro Incardona, Ata Deniz Aydin, Raimund Dachselt, Stefan Gumhold, Ivo F. Sbalzarini A Proposed Framework for Interactive Virtual Reality In Situ Visualization of Parallel Numerical Simulations.
In: 2019 IEEE 9th Symposium on Large Data Analysis and Visualization (LDAV)
(2019), Piscataway, N.J., IEEE (2019), 95-96
DOI
2018
Bevan Cheeseman, Ulrik Günther, Krzysztof Gonciarz, Mateusz Susik, Ivo F. Sbalzarini Adaptive particle representation of fluorescence microscopy images. Nat Commun, 9(1) Art. No. 5160 (2018)
Open AccessPDF
DOI
Modern microscopes create a data deluge with gigabytes of data generated each second, and terabytes per day. Storing and processing this data is a severe bottleneck, not fully alleviated by data compression. We argue that this is because images are processed as grids of pixels. To address this, we propose a content-adaptive representation of fluorescence microscopy images, the Adaptive Particle Representation (APR). The APR replaces pixels with particles positioned according to image content. The APR overcomes storage bottlenecks, as data compression does, but additionally overcomes memory and processing bottlenecks. Using noisy 3D images, we show that the APR adaptively represents the content of an image while maintaining image quality and that it enables orders of magnitude benefits across a range of image processing tasks. The APR provides a simple and efficient content-aware representation of fluosrescence microscopy images.
Dongcheng Zhang, James M Osborne, Kwaku Dad Abu-Bonsrah, Bevan Cheeseman, Kerry A Landman, Boaz Jurkowicz, Donald F Newgreen Stochastic clonal expansion of "superstars" enhances the reserve capacity of enteric nervous system precursor cells. Dev Biol, 444 Suppl 1 287-296 (2018)
DOI
We quantified cell population increase in the quail embryo enteric nervous system (ENS) from E2.5 (about 1500 cells) to E12 (about 8 million cells). We then probed ENS proliferative capacity by grafting to the chorio-allantoic membrane large (600 cells) and small (40 cells) populations of enteric neural crest (ENC) cells with aneural gut. This demonstrated that ENC cells show an extremely high capacity to regulate their proliferation while forming the ENS. Previous mathematical models and clonal label experiments revealed that a few dominant ENS "superstar" cell clones emerge but most clones are small. The model implied that "superstars" arise stochastically, but the same outcome could arise if "superstars" were pre-determined. We investigated these two modes mathematically and by grafting experiments with large and small numbers of ENCs, each including one EGFP-labelled ENC cell. The stochastic model predicts that the frequency of "superstar" detection increases as the ENC population decreases, the pre-determined model does not. Experimentally, as predicted by the stochastic model, the frequency of "superstar" detection increased with small ENC cell number. We conclude that ENS "superstar" clones achieve this status stochastically. Clonal dominance implies that clonal diversity is greatly reduced and in this case, somatic mutations may affect the phenotype. We suggest that somatic mutations coupled with loss of clonal diversity may contribute to variable penetrance and expressivity in individuals with genetically identical ENS pathologies.
Alexander Mietke Dynamics of active surfaces.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2018)
Michael Hecht, Ivo F. Sbalzarini Fast interpolation and Fourier transform in high-dimensional spaces.
In: Intelligent Computing : Proceedings of the 2018 IEEE Computing Conference, Vol. 2
(2018)(Eds.) Kohei Arai Advances in Intelligent Systems and Computing ; 857, Cham, Springer International Publishing (2018), 53-75
DOI
Michael Hecht Exact localisations of feedback sets Theory Comput Syst, 62(5) 1048-1084 (2018)
Open AccessPDF
DOI
Alejandro Vignoni, Anna Bajur, Elisabeth Knust, Ivo F. Sbalzarini Multi-objective identification from fluorescence recovery after photobleaching experiments: Understanding morphogenetic regulation of epithelial polarity IFAC-PapersOnLine, 51(19) 8-11 (2018)
Open Access
Ivo F. Sbalzarini, Urs F Greber How Computational Models Enable Mechanistic Insights into Virus Infection. Methods Mol Biol, 1836 609-631 (2018)
PDF
DOI
An implicit aim in cellular infection biology is to understand the mechanisms how viruses, microbes, eukaryotic parasites, and fungi usurp the functions of host cells and cause disease. Mechanistic insight is a deep understanding of the biophysical and biochemical processes that give rise to an observable phenomenon. It is typically subject to falsification, that is, it is accessible to experimentation and empirical data acquisition. This is different from logic and mathematics, which are not empirical, but built on systems of inherently consistent axioms. Here, we argue that modeling and computer simulation, combined with mechanistic insights, yields unprecedented deep understanding of phenomena in biology and especially in virus infections by providing a way of showing sufficiency of a hypothetical mechanism. This ideally complements the necessity statements accessible to empirical falsification by additional positive evidence. We discuss how computational implementations of mathematical models can assist and enhance the quantitative measurements of infection dynamics of enveloped and non-enveloped viruses and thereby help generating causal insights into virus infection biology.
Sven Karol, Tobias Nett, Jeronimo Castrillon, Ivo F. Sbalzarini A domain-specific language and editor for parallel particle methods. ACM Trans Math Softw, 44(3) Art. No. 34 (2018)
PDF
DOI
2017
Oleksandr Ostrenko, Pietro Incardona, Rajesh Ramaswamy, Lutz Brusch, Ivo F. Sbalzarini pSSAlib: The partial-propensity stochastic chemical network simulator. PLoS Comput Biol, 13(12) Art. No. e1005865 (2017)
Open Access DOI
Chemical reaction networks are ubiquitous in biology, and their dynamics is fundamentally stochastic. Here, we present the software library pSSAlib, which provides a complete and concise implementation of the most efficient partial-propensity methods for simulating exact stochastic chemical kinetics. pSSAlib can import models encoded in Systems Biology Markup Language, supports time delays in chemical reactions, and stochastic spatiotemporal reaction-diffusion systems. It also provides tools for statistical analysis of simulation results and supports multiple output formats. It has previously been used for studies of biochemical reaction pathways and to benchmark other stochastic simulation methods. Here, we describe pSSAlib in detail and apply it to a new model of the endocytic pathway in eukaryotic cells, leading to the discovery of a stochastic counterpart of the cut-out switch motif underlying early-to-late endosome conversion. pSSAlib is provided as a stand-alone command-line tool and as a developer API. We also provide a plug-in for the SBMLToolbox. The open-source code and pre-packaged installers are freely available from http://mosaic.mpi-cbg.de.
Bevan Cheeseman The Adaptive Particle Representation (APR) for Simple and Efficient Adaptive Resolution Processing, Storage and Simulations
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2017)
Yadira Boada, Alejandro Vignoni, Jesús Picó Multi-objective optimization for gene expression noise reduction in a synthetic gene circuit IFAC-PapersOnLine, 50(1) 4472-4477 (2017)
Open Access DOI
Stochasticity in biological systems often referred to as gene expression noise is ubiquitous. The main sources of this noise come about in two ways. The implicit randomness of the biochemical reactions generates intrinsic noise inside the cell. Other cellular processes are themselves products that vary over time and from each cell to another, producing the so-called extrinsic noise. Controlling the mean expression level of a gene while reducing its noise is a challenge in many applications of Synthetic Biology. In previous works, we proposed a gene synthetic circuit to reduce gene expression noise while achieving a desired mean expression level. The circuit combines a negative feedback loop and a cell-to-cell communication mechanism based on quorum sensing. In this work, we use a multi-objective optimization design approach to find the best values for the tunable-in-the-lab parameters that, for a given desired mean expression value, achieve minimization of gene expression noise caused by intrinsic and extrinsic fluctuations. Our approach allows tuning the circuit parameters required to minimize noise effects, providing results which prove in accordance with genome-wide experimental data reported in the literature. Exploring different scenarios, either considering only intrinsic noise or considering both extrinsic and intrinsic ones, we find that the design strategies obtained for both cases are not transferable. Thus, designing the circuit parameters taking into account only intrinsic noise yields a sub-optimal design with decreased performance when evaluated in a scenario where both extrinsic and intrinsic noise are present.
Yadira Boada, Alejandro Vignoni, Jesús Picó Engineered Control of Genetic Variability Reveals Interplay among Quorum Sensing, Feedback Regulation, and Biochemical Noise. ACS Synth Biol, 6(10) 1903-1912 (2017)
DOI
Stochastic fluctuations in gene expression trigger both beneficial and harmful consequences for cell behavior. Therefore, achieving a desired mean protein expression level while minimizing noise is of interest in many applications, including robust protein production systems in industrial biotechnology. Here, we consider a synthetic gene circuit combining intracellular negative feedback and cell-to-cell communication based on quorum sensing. Accounting for both intrinsic and extrinsic noise, stochastic simulations allow us to analyze the capability of the circuit to reduce noise strength as a function of its parameters. We obtain mean expression levels and noise strengths for all species under different scenarios, showing good agreement with system-wide available experimental data of protein abundance and noise in Escherichia coli. Our in silico experiments, validated by preliminary in vivo results, reveal significant noise attenuation in gene expression through the interplay between quorum sensing and negative feedback and highlight the differential role that they play in regard to intrinsic and extrinsic noise.
Maj Svea Grieb, Aleksandra Nivina, Bevan Cheeseman, Andreas Hartmann, Didier Mazel, Michael Schlierf Dynamic stepwise opening of integron attC DNA hairpins by SSB prevents toxicity and ensures functionality. Nucleic Acids Res, 45(18) 10555-10563 (2017)
Open Access DOI
Biologically functional DNA hairpins are found in archaea, prokaryotes and eukaryotes, playing essential roles in various DNA transactions. However, during DNA replication, hairpin formation can stall the polymerase and is therefore prevented by the single-stranded DNA binding protein (SSB). Here, we address the question how hairpins maintain their functional secondary structure despite SSB's presence. As a model hairpin, we used the recombinogenic form of the attC site, essential for capturing antibiotic-resistance genes in the integrons of bacteria. We found that attC hairpins have a conserved high GC-content near their apical loop that creates a dynamic equilibrium between attC fully opened by SSB and a partially structured attC-6-SSB complex. This complex is recognized by the recombinase IntI, which extrudes the hairpin upon binding while displacing SSB. We anticipate that this intriguing regulation mechanism using a base pair distribution to balance hairpin structure formation and genetic stability is key to the dissemination of antibiotic resistance genes among bacteria and might be conserved among other functional hairpins.
Karl Hoffmann, Anja Voss-Böhme, Jochen Rink, Lutz Brusch A dynamically diluted alignment model reveals the impact of cell turnover on the plasticity of tissue polarity patterns. J R Soc Interface, 14(135) Art. No. 20170466 (2017)
DOI
The polarization of cells and tissues is fundamental for tissue morphogenesis during biological development and regeneration. A deeper understanding of biological polarity pattern formation can be gained from the consideration of pattern reorganization in response to an opposing instructive cue, which we here consider using the example of experimentally inducible body axis inversions in planarian flatworms. We define a dynamically diluted alignment model linking three processes: entrainment of cell polarity by a global signal, local cell-cell coupling aligning polarity among neighbours, and cell turnover replacing polarized cells by initially unpolarized cells. We show that a persistent global orienting signal determines the final mean polarity orientation in this stochastic model. Combining numerical and analytical approaches, we find that neighbour coupling retards polarity pattern reorganization, whereas cell turnover accelerates it. We derive a formula for an effective neighbour coupling strength integrating both effects and find that the time of polarity reorganization depends linearly on this effective parameter and no abrupt transitions are observed. This allows us to determine neighbour coupling strengths from experimental observations. Our model is related to a dynamic 8-Potts model with annealed site-dilution and makes testable predictions regarding the polarization of dynamic systems, such as the planarian epithelium.
Sven Karol, Tobias Nett, Pietro Incardona, Nesrine Khouzami, Jeronimo Castrillon, Ivo F. Sbalzarini A Language and Development Environment for Parallel Particle Methods
In: V. International Conference on Particle-based Methods : Fundamentals and Applications ; PARTICLES 2017
(2017)(Eds.) Peter Wriggers, Barcelona, International Center for Numerical Methods in Engineering (CIMNE) (2017), 564-575
PDF
We present the Parallel Particle-Mesh Environment (PPME), a domainspecific language (DSL) and development environment for numerical simulations using particles and hybrid particle-mesh methods. PPME is the successor of the Parallel Particle-Mesh Language (PPML), a Fortran-based DSL that provides high-level abstractions for the development of distributed-memory particle-mesh simulations. On top of PPML, PPME provides a complete development environment for particle-based simulations usin state-of-the-art language engineering and compiler construction techniques. Relying on a novel domain metamodel and formal type system for particle methods, it enables advanced static code correctness checks at the level of particle abstractions, complementing the low-level analysis of the compiler. Furthermore, PPME adopts Herbie for improving the accuracy of floating-point expressions and supports a convenient high-level mathematical notation for equations and differential operators. For demonstration purposes, we discuss an example from Discrete Element Methods (DEM) using the classic Silbert model to simulate granular flows.
Josefine Asmus, Christian L Müller, Ivo F. Sbalzarini Lp-Adaptation: Simultaneous Design Centering and Robustness Estimation of Electronic and Biological Systems. Sci Rep, 7(1) Art. No. 6660 (2017)
Open AccessPDF
DOI
The design of systems or models that work robustly under uncertainty and environmental fluctuations is a key challenge in both engineering and science. This is formalized in the design-centering problem, which is defined as finding a design that fulfills given specifications and has a high probability of still doing so if the system parameters or the specifications fluctuate randomly. Design centering is often accompanied by the problem of quantifying the robustness of a system. Here we present a novel adaptive statistical method to simultaneously address both problems. Our method, L p -Adaptation, is inspired by the evolution of robustness in biological systems and by randomized schemes for convex volume computation. It is able to address both problems in the general, non-convex case and at low computational cost. We describe the concept and the algorithm, test it on known benchmarks, and demonstrate its real-world applicability in electronic and biological systems. In all cases, the present method outperforms the previous state of the art. This enables re-formulating optimization problems in engineering and biology as design centering problems, taking global system robustness into account.
Yadira Boada, Alejandro Vignoni, Jesús Picó Multi-objective identification of synthetic circuits stochastic models using flow flcytometry data
In: 2017 25TH MEDITERRANEAN CONFERENCE ON CONTROL AND AUTOMATION (MED) : July 3-6, 2017, University of Malta, Valetta Campus, Malta
(2017) Mediterranean Conference on Control and Automation, Piscataway, N.J., IEEE (2017), 1077-1082
DOI
Gerald Hempel, Goens Andres, Josefine Asmus, Jeronimo Castrillon, Ivo F. Sbalzarini Robust Mapping of Process Networks to Many-Core Systems Using Bio-Inspired Design Centering
In: Proceedings of the 20th International Workshop on Software and Compilers for Embedded Systems - SCOPES '17
(2017)(Eds.) Sander Stuijk, New York, ACM (2017), 21-30
PDF
Yuanhao Gong, Ivo F. Sbalzarini Curvature Filters Efficiently Reduce Certain Variational Energies. IEEE Trans Image Process, 26(4) 1786-1798 (2017)
Open AccessPDF
DOI
In image processing, the rapid approximate solution of variational problems involving generic data-fitting terms is often of practical relevance, for example in real-time applications. Variational solvers based on diffusion schemes or the Euler-Lagrange equations are too slow and restricted in the types of data-fitting terms. Here, we present a filter-based approach to reduce variational energies that contain generic data-fitting terms, but are restricted to specific regularizations. Our approach is based on reducing the regularization part of the variational energy, while guaranteeing non-increasing total energy. This is applicable to regularization-dominated models, where the data-fitting energy initially increases, while the regularization energy initially decreases. We present fast discrete filters for regularizers based on Gaussian curvature, mean curvature, and total variation. These pixel-local filters can be used to rapidly reduce the energy of the full model. We prove the convergence of the resulting iterative scheme in a greedy sense, and we show several experiments to demonstrate applications in image-processing problems involving regularization-dominated variational models.
Kirstin Meyer, Oleksandr Ostrenko, George Bourantas, Hernán Morales-Navarrete, Natalie Porat-Shliom, Fabián Segovia-Miranda, Hidenori Nonaka, Ali Ghaemi, Jean-Marc Verbavatz, Lutz Brusch, Ivo F. Sbalzarini, Yannis Kalaidzidis, Roberto Weigert, Marino Zerial A Predictive 3D Multi-Scale Model of Biliary Fluid Dynamics in the Liver Lobule. Cell Syst, 4(3) 277-290 (2017)
Open AccessPDF
DOI
Bile, the central metabolic product of the liver, is transported by the bile canaliculi network. The impairment of bile flow in cholestatic liver diseases has urged a demand for insights into its regulation. Here, we developed a predictive 3D multi-scale model that simulates fluid dynamic properties successively from the subcellular to the tissue level. The model integrates the structure of the bile canalicular network in the mouse liver lobule, as determined by high-resolution confocal and serial block-face scanning electron microscopy, with measurements of bile transport by intravital microscopy. The combined experiment-theory approach revealed spatial heterogeneities of biliary geometry and hepatocyte transport activity. Based on this, our model predicts gradients of bile velocity and pressure in the liver lobule. Validation of the model predictions by pharmacological inhibition of Rho kinase demonstrated a requirement of canaliculi contractility for bile flow in vivo. Our model can be applied to functionally characterize liver diseases and quantitatively estimate biliary transport upon drug-induced liver injury.
Donald F Newgreen, Dongcheng Zhang, Bevan Cheeseman, Benjamin J Binder, Kerry A Landman Differential Clonal Expansion in an Invading Cell Population: Clonal Advantage or Dumb Luck? Cells Tissues Organs , 203(2) 105-113 (2017)
DOI
In neoplastic cell growth, clones and subclones are variable both in size and mutational spectrum. The largest of these clones are believed to represent those cells with mutations that make them the most "fit," in a Darwinian sense, for expansion in their microenvironment. Thus, the degree of quantitative clonal expansion is regarded as being determined by innate qualitative differences between the cells that originate each clone. Here, using a combination of mathematical modelling and clonal labelling experiments applied to the developmental model system of the forming enteric nervous system, we describe how cells which are qualitatively identical may consistently produce clones of dramatically different sizes: most clones are very small while a few clones we term "superstars" contribute most of the cells to the final population. The basis of this is minor stochastic variations ("luck") in the timing and direction of movement and proliferation of individual cells, which builds a local advantage for daughter cells that is cumulative. This has potentially important consequences. In cancers, especially before strongly selective cytotoxic therapy, the assumption that the largest clones must be the cells with deterministic proliferative ability may not always hold true. In development, the gradual loss of clonal diversity as "superstars" take over the population may erode the resilience of the system to somatic mutations, which may have occurred early in clonal growth.
Josefine Asmus An efficient randomized approximation algorithm for volume estimation and design centering
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2017) PDF
2016
Yadira Boada, Alejandro Vignoni, Gilberto Reynoso-Meza, Jesús Picó Parameter identification in synthetic biological circuits using multi-objective optimization IFAC-PapersOnLine, 49(26) 77-82 (2016)
Open AccessPDF
DOI
Enric Picó-Marco, Yadira Boada, Jesús Picó, Alejandro Vignoni Contractivity of a genetic circuit with internal feedback and cell-to-cell communication IFAC-PapersOnLine, 49(26) 213-218 (2016)
Open AccessPDF
DOI
Yaser Afshar Parallel distributed-memory particle methods for acquisition-rate segmentation and uncertainty quantifications of large fluorescence microscopy images.
Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany (2016) PDF
George Bourantas*, Bevan Cheeseman*, Rajesh Ramaswamy, Ivo F. Sbalzarini Using DC PSE operator discretization in Eulerian meshless collocation methods improves their robustness in complex geometries Computers & Fluids, 136 285-300 (2016)
Open AccessPDF
DOI
Ivo F. Sbalzarini Seeing Is Believing: Quantifying Is Convincing: Computational Image Analysis in Biology
In: Focus on Bio-Image Informatics. (Eds.) Winnok, H. De Vos (Advances in Anatomy, Embryology and Cell Biology ; 219)., Cham, Springer International Publishing (2016), 1-39 Ch. 1 PDF
DOI
Janko Kajtez, Anastasia Solomatina, Maja Novak, Bruno Polak, Kruno Vukušić, Jonas Rüdiger, Gheorghe Cojoc, Ana Milas, Ivana Šumanovac Šestak, Patrik Risteski, Federica Tavano, Anna Klemm, Emanuele Roscioli, Julie Welburn, Daniela Cimini, Matko Glunčić, Nenad Pavin, Iva M Tolić Overlap microtubules link sister k-fibres and balance the forces on bi-oriented kinetochores. Nat Commun, 7 Art. No. 10298 (2016)
Open AccessPDF
DOI
During metaphase, forces on kinetochores are exerted by k-fibres, bundles of microtubules that end at the kinetochore. Interestingly, non-kinetochore microtubules have been observed between sister kinetochores, but their function is unknown. Here we show by laser-cutting of a k-fibre in HeLa and PtK1 cells that a bundle of non-kinetochore microtubules, which we term 'bridging fibre', bridges sister k-fibres and balances the interkinetochore tension. We found PRC1 and EB3 in the bridging fibre, suggesting that it consists of antiparallel dynamic microtubules. By using a theoretical model that includes a bridging fibre, we show that the forces at the pole and at the kinetochore depend on the bridging fibre thickness. Moreover, our theory and experiments show larger relaxation of the interkinetochore distance for cuts closer to kinetochores. We conclude that the bridging fibre, by linking sister k-fibres, withstands the tension between sister kinetochores and enables the spindle to obtain a curved shape.
Yadira Boada, José Luis Pitarch, Alejandro Vignoni, Gilberto Reynoso-Meza, Jesús Picó Optimization Alternatives for Robust Model-based Design of Synthetic Biological Circuits IFAC-PapersOnLine, 49(7) 821-826 (2016)
Open AccessPDF
DOI
Franck Raynaud, Mark E. Ambühl, Chiara Gabella, Alicia Bornert, Ivo F. Sbalzarini, J-J Meister, A B Verkhovsky Minimal model for spontaneous cell polarization and edge activity in oscillating, rotating and migrating cells. Nat Phys, 12 367-373 (2016)
DOI
Artur Yakimovich, Yauhen Yakimovich, Michael Schmid, Jason Mercer, Ivo F. Sbalzarini, Urs F Greber Infectio: a Generic Framework for Computational Simulation of Virus Transmission between Cells. mSphere, 1(1) Art. No. e00078-15 (2016)
Open AccessPDF
DOI
Viruses spread between cells, tissues, and organisms by cell-free and cell-cell mechanisms, depending on the cell type, the nature of the virus, or the phase of the infection cycle. The mode of viral transmission has a large impact on disease development, the outcome of antiviral therapies or the efficacy of gene therapy protocols. The transmission mode of viruses can be addressed in tissue culture systems using live-cell imaging. Yet even in relatively simple cell cultures, the mechanisms of viral transmission are difficult to distinguish. Here we present a cross-platform software framework called "Infectio," which is capable of simulating transmission phenotypes in tissue culture of virtually any virus. Infectio can estimate interdependent biological parameters, for example for vaccinia virus infection, and differentiate between cell-cell and cell-free virus spreading. Infectio assists in elucidating virus transmission mechanisms, a feature useful for designing strategies of perturbing or enhancing viral transmission. The complexity of the Infectio software is low compared to that of other software commonly used to quantitate features of cell biological images, which yields stable and relatively error-free output from Infectio. The software is open source (GPLv3 license), and operates on the major platforms (Windows, Mac, and Linux). The complete source code can be downloaded from http://infectio.github.io/index.html. IMPORTANCE Infectio presents a generalized platform to analyze virus infection spread between cells. It allows the simulation of plaque phenotypes from image-based assays. Viral plaques are the result of virus spreading from primary infected cells to neighboring cells. This is a complex process and involves neighborhood effects at cell-cell contact sites or fluid dynamics in the extracellular medium. Infectio differentiates between two major modes of virus transmission between cells, allowing in silico testing of hypotheses about spreading mechanisms of any virus which can be grown in cell cultures, based on experimentally measured parameters, such as infection intensity or cell killing. The results of these tests can be compared with experimental data and allow interpretations with regard to biophysical mechanisms. Infectio also facilitates characterizations of the mode of action of therapeutic agents, such as oncolytic viruses or other infectious or cytotoxic agents.
Xun Xiao, Veikko Geyer, Hugo Bowne-Anderson, Jonathon Howard, Ivo F. Sbalzarini Automatic optimal filament segmentation with sub-pixel accuracy using generalized linear models and B-spline level-sets Med Image Anal, 32 157-172 (2016)
Open AccessPDF
DOI
Yaser Afshar, Ivo F. Sbalzarini A Parallel Distributed-Memory Particle Method Enables Acquisition-Rate Segmentation of Large Fluorescence Microscopy Images PLoS ONE, 11(4) Art. No. e0152528 (2016)
Open AccessPDF
DOI
Yuanhao Gong, Ivo F. Sbalzarini A Natural-Scene Gradient Distribution Prior and its Application in Light-Microscopy Image Processing IEEE J Sel Top Signal Process, 10(1) 99-114 (2016)
Open AccessPDF
2015
Sven Karol, Pietro Incardona, Yaser Afshar, Ivo F. Sbalzarini, Jeronimo Castrillon Towards a Next-Generation Parallel Particle-Mesh Language.
In: Proceedings of the 3rd Workshop on Domain-Specific Language Design and Implementation DSLDI’15 at 29th European Conference on Object-Oriented Programming (ECOOP 2015), Prague, Czech Republic, July 7, 2015
(2015)(Eds.) Sebastian Erdweg, Tijs van der Storm, Cornell University Library, arXiv (2015)
PDF
Loic Royer, Martin Weigert, Ulrik Günther, Nicola Maghelli, Florian Jug, Ivo F. Sbalzarini, Eugene W Myers ClearVolume: open-source live 3D visualization for light-sheet microscopy. Nat Methods, 12(6) 480-481 (2015)
PDF
DOI
Yuanhao Gong, Ivo F. Sbalzarini Image Enhancement by Gradient Distribution Specification
In: Proceedings ACCV, 12th Asian Conference on Computer Vision, Workshop on Emerging Topics in Image Enhancement and Restoration
(2015) Lecture Notes in Computer Science, New York, Springer (2015), 47-62
PDF
Yuanhao Gong Spectrally Regularized Surfaces
Ph.D. Thesis, ETH Zurich, Department of Computer Science, Zurich, Switzerland (2015)
Christoph Häcki, Sylvain Reboux, Ivo F. Sbalzarini A Self-Organizing Adaptive-Resolution Particle Methods with Anisotropic Kernels Proc IUTAM, 18 40-55 (2015)
Open AccessPDF
Rajesh Ramaswamy, George Bourantas, Frank Jülicher, Ivo F. Sbalzarini A hybrid particle-mesh method for incompressible active polar viscous gels J Comput Phys, 291 334-361 (2015)
Open AccessPDF
2014
Ömer Demirel, Ihor Smal, Wiro J. Niessen, Erik Meijering, Ivo F. Sbalzarini An adaptive distributed resampling algorithm with non-proportional allocation
In: IEEE International Conference on Acoustics, Speech, and Signal Processing (ICASSP) : Florence, Italy, 4-9 May 2014
(2014), Piscataway, N.J., IEEE (2014), 1635-1639
PDF
Aurelien Rizk, Gregory Paul, Pietro Incardona, Milica Bugarski, Maysam Mansouri, Axel Niemann, Urs Ziegler, Philipp Berger#, Ivo F. Sbalzarini# Segmentation and quantification of subcellular structures in fluorescence microscopy images using Squassh. Nat Protoc, 9(3) 586-596 (2014)
PDF
DOI
Detection and quantification of fluorescently labeled molecules in subcellular compartments is a key step in the analysis of many cell biological processes. Pixel-wise colocalization analyses, however, are not always suitable, because they do not provide object-specific information, and they are vulnerable to noise and background fluorescence. Here we present a versatile protocol for a method named 'Squassh' (segmentation and quantification of subcellular shapes), which is used for detecting, delineating and quantifying subcellular structures in fluorescence microscopy images. The workflow is implemented in freely available, user-friendly software. It works on both 2D and 3D images, accounts for the microscope optics and for uneven image background, computes cell masks and provides subpixel accuracy. The Squassh software enables both colocalization and shape analyses. The protocol can be applied in batch, on desktop computers or computer clusters, and it usually requires <1 min and <5 min for 2D and 3D images, respectively. Basic computer-user skills and some experience with fluorescence microscopy are recommended to successfully use the protocol.
Josefine Asmus, Daniel Borchmann, Ivo F. Sbalzarini, Dirk Walther Towards an FCA-based Recommender System for Black-Box Optimization
In: Proceedings of the 3rd International Workshop "What can FCA do for Artificial Intelligence"? co-located with the European Conference on Artificial Intelligence (ECAI 2014) Prague, Czech Republic, August 19, 2014
(2014), Aachen, CEUR Workshop Proceedings (2014), 35-42
PDF
Christian Bläsche Setting Young’s modulus in the subcellular element model
Diploma Thesis,Technische Universität Dresden, Dresden, Germany (2014) PDF
Nicolas Chenouard, Ihor Smal, Fabrice de Chaumont, Martin Maška, Ivo F. Sbalzarini, Yuanhao Gong, Janick Cardinale, Craig Carthel, Stefano Coraluppi, Mark Winter, Andrew R Cohen, William J. Godinez, Karl Rohr, Yannis Kalaidzidis, Liang Liang, James Duncan, Hongying Shen, Yingke Xu, Klas E. G. Magnusson, Joakim Jaldén, Helen M. Blau, Perrine Paul-Gilloteaux, Philippe Roudot, Charles Kervrann, Francois Waharte, Jean-Yves Tinevez, Spencer L Shorte, Joost Willemse, Katherine Celler, Gilles P van Wezel, Han-Wei Dan, Yuh-Show Tsai, Carlos Ortiz de Solórzano, Jean-Christophe Olivo-Marin#, Erik Meijering# Objective comparison of particle tracking methods. Nat Methods, 11(3) 281-289 (2014)
PDF
DOI
Particle tracking is of key importance for quantitative analysis of intracellular dynamic processes from time-lapse microscopy image data. Because manually detecting and following large numbers of individual particles is not feasible, automated computational methods have been developed for these tasks by many groups. Aiming to perform an objective comparison of methods, we gathered the community and organized an open competition in which participating teams applied their own methods independently to a commonly defined data set including diverse scenarios. Performance was assessed using commonly defined measures. Although no single method performed best across all scenarios, the results revealed clear differences between the various approaches, leading to notable practical conclusions for users and developers.
Ömer Demirel Dynamic Load Balancing in Parallel Particle Methods
Ph.D. Thesis, ETH Zurich, Department of Computer Science, Zurich, Switzerland (2014) PDF
Chiara Gabella, Elena Bertseva, Céline Bottier, Niccolo Piacentini, Alicia Bornert, Sylvia Jeney, Laszlo Forro, Ivo F. Sbalzarini, J-J Meister, A B Verkhovsky Contact Angle at the Leading Edge Controls Cell Protrusion Rate Curr Biol, 24(10) 1126-1132 (2014)
PDF
DOI
Fabian Multrus Calculation of the Electric Potential for a Neuronal Activity Model in the Brain
Diploma Thesis,Technische Universität Dresden, Dresden, Germany (2014) PDF
Omar Awile, Ivo F. Sbalzarini A pthreads wrapper for Fortran 2003 ACM Trans Math Softw, 40(3) Art. No. 19 (2014)
PDF
DOI
Yuri K. Shestopaloff, Ivo F. Sbalzarini A method for modeling growth of organs and transplants based on the general growth law: application to the liver in dogs and humans. PLoS ONE, 9(6) Art. No. e99275 (2014)
Open Access DOI
Understanding biological phenomena requires a systemic approach that incorporates different mechanisms acting on different spatial and temporal scales, since in organisms the workings of all components, such as organelles, cells, and organs interrelate. This inherent interdependency between diverse biological mechanisms, both on the same and on different scales, provides the functioning of an organism capable of maintaining homeostasis and physiological stability through numerous feedback loops. Thus, developing models of organisms and their constituents should be done within the overall systemic context of the studied phenomena. We introduce such a method for modeling growth and regeneration of livers at the organ scale, considering it a part of the overall multi-scale biochemical and biophysical processes of an organism. Our method is based on the earlier discovered general growth law, postulating that any biological growth process comprises a uniquely defined distribution of nutritional resources between maintenance needs and biomass production. Based on this law, we introduce a liver growth model that allows to accurately predicting the growth of liver transplants in dogs and liver grafts in humans. Using this model, we find quantitative growth characteristics, such as the time point when the transition period after surgery is over and the liver resumes normal growth, rates at which hepatocytes are involved in proliferation, etc. We then use the model to determine and quantify otherwise unobservable metabolic properties of livers.
2013
Ivo F. Sbalzarini Modeling and simulation of biological systems from image data. Bioessays, 35(5) 482-490 (2013)
PDF
DOI
This essay provides an introduction to the terminology, concepts, methods, and challenges of image-based modeling in biology. Image-based modeling and simulation aims at using systematic, quantitative image data to build predictive models of biological systems that can be simulated with a computer. This allows one to disentangle molecular mechanisms from effects of shape and geometry. Questions like "what is the functional role of shape" or "how are biological shapes generated and regulated" can be addressed in the framework of image-based systems biology. The combination of image quantification, model building, and computer simulation is illustrated here using the example of diffusion in the endoplasmic reticulum.
Ferit Büyükkeçeci, Omar Awile, Ivo F. Sbalzarini A portable OpenCL implementation of generic particle-mesh and mesh-particle interpolation 2D and 3D Parallel Comput, 39(2) 94-111 (2013)
PDF
DOI
Janick Cardinale Unsupervised Segmentation and Shape Posterior Estimation under Bayesian Image Models
Ph.D. Thesis, ETH Zurich, Department of Computer Science, Zurich, Switzerland (2013) PDF
Oliwia M. Szklarczyk, Nélido González-Segredo, Philipp Kukura, Ariella Oppenheim, Daniel Choquet, Vahid Sandoghdar, Ari Helenius, Ivo F. Sbalzarini, Helge Ewers Receptor Concentration and Diffusivity Control Multivalent Binding of SV40 to Membrane Bilayers PLoS Comput Biol, 9(11) Art. No. e1003310 (2013)
PDF
Katharina Philipp Piecewise smooth deconvolving segmentation
Diploma Thesis,Technische Universität Dresden, Dresden, Germany (2013) PDF
Ali Ghaemi Numerical simulations of bile flow in realistic image-derived bile canalicular geometries
Diploma Thesis,Technische Universität Dresden, Dresden, Germany (2013) PDF
Arun Shivanandan, Aleksandra Radenovic, Ivo F. Sbalzarini MosaicIA: an ImageJ/Fiji plugin for spatial pattern and interaction analysis BMC Bioinformatics, 14 Art. No. 349 (2013)
Open AccessPDF
Yuanhao Gong, Ivo F. Sbalzarini Local weighted Gaussian curvature for image processing
In: 2013 IEEE International Conference on Image Processing (ICIP 2013) : Melbourne, Australia, 15 - 18 September
(2013), Piscataway, N.J., IEEE (2013), 534-538
PDF
Gregory Paul, Janick Cardinale, Ivo F. Sbalzarini Coupling Image Restoration and Segmentation: A Generalized Linear Model/Bregman Perspective Int J Comput Vis, 104(1) 69-93 (2013)
PDF
DOI
Nicolas Fietier, Ömer Demirel, Ivo F. Sbalzarini A meshless particle method for Poisson and diffusion problems with discontinuous coefficients and inhomogeneous boundary conditions SIAM J Sci Comput, 35(6) 2469-2493 (2013)
Omar Awile, Milan Mitrovic, Sylvain Reboux, Ivo F. Sbalzarini A domain-specific programming language for particle simulations on distributed-memory parallel computers
In: Particle-based methods III : fundamentals and applications ; proceedings of the III International Conference on Particle-Based Methods, Fundamentals and Applications (Particles 2013), Stuttgart, 18 - 20 September
(2013), Barcelona, International Center for Numerical Methods in Engineering (CIMNE) (2013), 436-447
PDF
Omar Awile A Domain-Specific Language and Scalable Middleware for Particle-Mesh Simulations on Heterogeneous Parallel Computers
Ph.D. Thesis, ETH Zurich, Department of Computer Science, Zurich, Switzerland (2013) PDF
2012
Artur Yakimovich, Heidi Gumpert, C J Burckhardt, Verena A Lütschg, Andreas Jurgeit, Ivo F. Sbalzarini, U F Greber Cell-free transmission of human adenovirus by passive mass transfer in cell culture simulated in a computer model. J Virol, 86(18) 10123-10137 (2012)
PDF
DOI
Viruses spread between cells, tissues, and organisms by cell-free and cell-cell transmissions. Both mechanisms enhance disease development, but it is difficult to distinguish between them. Here, we analyzed the transmission mode of human adenovirus (HAdV) in monolayers of epithelial cells by wet laboratory experimentation and a computer simulation. Using live-cell fluorescence microscopy and replication-competent HAdV2 expressing green fluorescent protein, we found that the spread of infection invariably occurred after cell lysis. It was affected by convection and blocked by neutralizing antibodies but was independent of second-round infections. If cells were overlaid with agarose, convection was blocked and round plaques developed around lytic infected cells. Infected cells that did not lyse did not give rise to plaques, highlighting the importance of cell-free transmission. Key parameters for cell-free virus transmission were the time from infection to lysis, the dose of free viruses determining infection probability, and the diffusion of single HAdV particles in aqueous medium. With these parameters, we developed an in silico model using multiscale hybrid dynamics, cellular automata, and particle strength exchange. This so-called white box model is based on experimentally determined parameters and reproduces viral infection spreading as a function of the local concentration of free viruses. These analyses imply that the extent of lytic infections can be determined by either direct plaque assays or can be predicted by calculations of virus diffusion constants and modeling.
Janick Cardinale, Gregory Paul, Ivo F. Sbalzarini Discrete region competition for unknown numbers of connected regions IEEE Trans Image Process, 21(8) 3531-3545 (2012)
PDF
DOI
We present a discrete, unsupervised multi-regioncompetition algorithm for image segmentation over different energy functionals. The number of regions present in an image does not need to be known a priori, nor their photometric properties. The algorithm jointly estimates the number of regions, their photometries, and their contours. The required regularization is provided by defining a region as a connected set of pixels. The evolving contours in the image are represented by computational particles that move as driven by an energy-minimization algorithm. We present an efficient discrete algorithm that allows minimizing a range of well-known energy functionals under the topological constraint of regions being connected components. The presented framework and algorithms are implemented in the open-source Insight Toolkit (ITK) image-processing library.
Ivo F. Sbalzarini Active flows cluster cell surface proteins. Dev Cell, 22(6) 1121-1122 (2012)
PDF
DOI
The plasma membrane of cells is a dynamic mixture of different lipids, proteins, and sugars. In a recent issue of Cell, Gowrishankar et al. (2012) propose a model for how the actin cortex may generate and regulate lateral heterogeneity in the plasma membrane by actively clustering cell surface molecules.
Sylvain Reboux, Birte Schrader, Ivo F. Sbalzarini A self-organizing Lagrangian particle method for adaptive-resolution advection–diffusion simulations J Comput Phys, 231(9) 3623-3646 (2012)
PDF
Mark E. Ambühl, C Brepsant, J-J Meister, A B Verkhovsky, Ivo F. Sbalzarini High-resolution cell outline segmentation and tracking from phase-contrast microscopy images. J Microsc, 245(2) 161-170 (2012)
PDF
DOI
Accurate extraction of cell outlines from microscopy images is essential for analysing the dynamics of migrating cells. Phase-contrast microscopy is one of the most common and convenient imaging modalities for observing cell motility because it does not require exogenous labelling and uses only moderate light levels with generally negligible phototoxicity effects. Automatic extraction and tracking of high-resolution cell outlines from phase-contrast images, however, is difficult due to complex and non-uniform edge intensity. We present a novel image-processing method based on refined level-set segmentation for accurate extraction of cell outlines from high-resolution phase-contrast images. The algorithm is validated on synthetic images of defined noise levels and applied to real image sequences of polarizing and persistently migrating keratocyte cells. We demonstrate that the algorithm is able to reliably reveal fine features in the cell edge dynamics.
Sebastian Stich, Christian L. Müller On spectral invariance of Randomized Hessian and Covariance Matrix Adaptation Schemes
In: Proc. Parallel Problem Solving From Nature (PPSN)
(2012) Lecture Notes in Computer Science, Berlin;Heidelberg, Springer (2012), 1-1
PDF
Christian L. Müller, Rajesh Ramaswamy, Ivo F. Sbalzarini Global parameter identification of stochastic reaction networks from single trajectories. Adv Exp Med Biol, 736 477-498 (2012)
PDF
DOI
We consider the problem of inferring the unknown parameters of a stochastic biochemical network model from a single measured time-course of the concentration of some of the involved species. Such measurements are available, e.g., from live-cell fluorescence microscopy in image-based systems biology. In addition, fluctuation time-courses from, e.g., fluorescence correlation spectroscopy (FCS) provide additional information about the system dynamics that can be used to more robustly infer parameters than when considering only mean concentrations. Estimating model parameters from a single experimental trajectory enables single-cell measurements and quantification of cell-cell variability. We propose a novel combination of an adaptive Monte Carlo sampler, called Gaussian Adaptation (GaA), and efficient exact stochastic simulation algorithms (SSA) that allows parameter identification from single stochastic trajectories. We benchmark the proposed method on a linear and a non-linear reaction network at steady state and during transient phases. In addition, we demonstrate that the present method also provides an ellipsoidal volume estimate of the viable part of parameter space and is able to estimate the physical volume of the compartment in which the observed reactions take place.
Omar Awile, Ferit Büyükkeçeci, Sylvain Reboux, Ivo F. Sbalzarini Fast neighbor lists for adaptive-resolution particle simulations Comput Phys Commun, 183(5) 1073-1081 (2012)
PDF
Christian L. Müller, Ivo F. Sbalzarini Energy landscapes of atomic clusters as black box optimization benchmarks. Evol Comput, 20(4) 543-573 (2012)
PDF
DOI
We present the energy minimization of atomic clusters as a promising problem class for continuous black box optimization benchmarks. Finding the arrangement of atoms that minimizes a given potential energy is a specific instance of the more general class of geometry optimization or packing problems, which are generally NP-complete. Atomic clusters are a well-studied subject in physics and chemistry. From the large set of available cluster optimization problems, we propose two specific instances: Cohn-Kumar clusters and Lennard-Jones clusters. The potential energies of these clusters are governed by distance-dependent pairwise interaction potentials. The resulting collection of landscapes is composed of smooth and rugged single-funnel topologies, as well as tunable double-funnel topologies. In addition, all problems possess a feature that is not covered by the synthetic functions in current black box optimization test suites: isospectral symmetry. This property implies that any atomic arrangement is uniquely defined by the pairwise distance spectrum, rather than the absolute atomic positions. We hence suggest that the presented problem instances should be included in black box optimization benchmark suites.
Rajesh Ramaswamy, Nélido González-Segredo, Ivo F. Sbalzarini, Ramon Grima Discreteness-induced concentration inversion in mesoscopic chemical systems Nat Commun, 3 Art. No. 779 (2012)
PDF
DOI
Molecular discreteness is apparent in small-volume chemical systems, such as biological cells, leading to stochastic kinetics. Here we present a theoretical framework to understand the effects of discreteness on the steady state of a monostable chemical reaction network. We consider independent realizations of the same chemical system in compartments of different volumes. Rate equations ignore molecular discreteness and predict the same average steady-state concentrations in all compartments. However, our theory predicts that the average steady state of the system varies with volume: if a species is more abundant than another for large volumes, then the reverse occurs for volumes below a critical value, leading to a concentration inversion effect. The addition of extrinsic noise increases the size of the critical volume. We theoretically predict the critical volumes and verify, by exact stochastic simulations, that rate equations are qualitatively incorrect in sub-critical volumes.
Yuanhao Gong, Gregory Paul, Ivo F. Sbalzarini Coupled signed-distance functions for implicit surface reconstruction
In: Proc. IEEE Intl. Symposium Biomedical Imaging (ISBI)
(2012), Piscataway, N.J., IEEE (2012), 1000-1003
PDF
Birte Schrader, Sylvain Reboux, Ivo F. Sbalzarini Choosing the best kernel: performance models for diffusion operators in particle methods SIAM J Sci Comput, 34(3) 1607-1634 (2012)
PDF
DOI
2011
Jo A. Helmuth, Sylvain Reboux, Ivo F. Sbalzarini Exact stochastic simulations of intra-cellular transport by mechanically coupled molecular motors J Comput Sci, 2(4) 324-334 (2011)
PDF
Rajesh Ramaswamy, Ivo F. Sbalzarini Exact on-lattice stochastic reaction-diffusion simulations using partial-propensity methods J Chem Phys, 135(24) Art. No. 244103 (2011)
PDF
DOI
Stochastic reaction-diffusion systems frequently exhibit behavior that is not predicted by deterministic simulation models. Stochastic simulation methods, however, are computationally expensive. We present a more efficient stochastic reaction-diffusion simulation algorithm that samples realizations from the exact solution of the reaction-diffusion master equation. The present algorithm, called partial-propensity stochastic reaction-diffusion (PSRD) method, uses an on-lattice discretization of the reaction-diffusion system and relies on partial-propensity methods for computational efficiency. We describe the algorithm in detail, provide a theoretical analysis of its computational cost, and demonstrate its computational performance in benchmarks. We then illustrate the application of PSRD to two- and three-dimensional pattern-forming Gray-Scott systems, highlighting the role of intrinsic noise in these systems.
Céline Bottier, Chiara Gabella, Benoît Vianay, Lara Buscemi, Ivo F. Sbalzarini, J-J Meister, A B Verkhovsky Dynamic measurement of the height and volume of migrating cells by a novel fluorescence microscopy technique. Lab on a chip, 11(22) 3855-3863 (2011)
PDF
DOI
We propose a new technique to measure the volume of adherent migrating cells. The method is based on a negative staining where a fluorescent, non-cell-permeant dye is added to the extracellular medium. The specimen is observed with a conventional fluorescence microscope in a chamber of uniform height. Given that the fluorescence signal depends on the thickness of the emitting layer, the objects excluding the fluorescent dye (i.e., cells) appear dark, and the decrease of the fluorescent signal with respect to the background is expected to give information about the height and the volume of the object. Using a glass microfabricated pattern with steps of defined heights, we show that the drop in fluorescence intensity is indeed proportional to the height of the step and obtain calibration curves relating fluorescence intensity to height. The technique, termed the fluorescence displacement method, is further validated by comparing our measurements with the ones obtained by atomic force microscopy (AFM). We apply our method to measure the real-time volume dynamics of migrating fish epidermal keratocytes subjected to osmotic stress. The fluorescence displacement technique allows fast and precise monitoring of cell height and volume, thus providing a valuable tool for characterizing the three-dimensional behaviour of migrating cells.
Yohei Yamauchi, Heithem Boukari, Indranil Banerjee, Ivo F. Sbalzarini, Peter Horvath, Ari Helenius Histone deacetylase 8 is required for centrosome cohesion and influenza A virus entry PLoS Pathog, 7(10) 1002316-1002316 (2011)
PDF
DOI
Influenza A virus (IAV) enters host cells by endocytosis followed by acid-activated penetration from late endosomes (LEs). Using siRNA silencing, we found that histone deacetylase 8 (HDAC8), a cytoplasmic enzyme, efficiently promoted productive entry of IAV into tissue culture cells, whereas HDAC1 suppressed it. HDAC8 enhanced endocytosis, acidification, and penetration of the incoming virus. In contrast, HDAC1 inhibited acidification and penetration. The effects were connected with dramatic alterations in the organization of the microtubule system, and, as a consequence, a change in the behavior of LEs and lysosomes (LYs). Depletion of HDAC8 caused loss of centrosome-associated microtubules and loss of directed centripetal movement of LEs, dispersing LE/LYs to the cell periphery. For HDAC1, the picture was the opposite. To explain these changes, centrosome cohesion emerged as the critical factor. Depletion of HDAC8 caused centrosome splitting, which could also be induced by depleting a centriole-linker protein, rootletin. In both cases, IAV infection was inhibited. HDAC1 depletion reduced the splitting of centrosomes, and enhanced infection. The longer the distance between centrosomes, the lower the level of infection. HDAC8 depletion was also found to inhibit infection of Uukuniemi virus (a bunyavirus) suggesting common requirements among late penetrating enveloped viruses. The results established class I HDACs as powerful regulators of microtubule organization, centrosome function, endosome maturation, and infection by IAV and other late penetrating viruses.
Gregory Paul, Christian L. Müller, Ivo F. Sbalzarini Sensitivity analysis from evolutionary algorithm search paths
In: Proc. EVOLVE
(2011), Luxembourg, EVOLVE (2011), 1-4
PDF
Zlatko Smole, Nela Nikolic, Fran Supek, Tomislav Šmuc, Ivo F. Sbalzarini, Anita Krisko Proteome sequence features carry signatures of the environmental niche of prokaryotes. BMC Evol Biol, 11 26-26 (2011)
Open AccessPDF
DOI
Prokaryotic environmental adaptations occur at different levels within cells to ensure the preservation of genome integrity, proper protein folding and function as well as membrane fluidity. Although specific composition and structure of cellular components suitable for the variety of extreme conditions has already been postulated, a systematic study describing such adaptations has not yet been performed. We therefore explored whether the environmental niche of a prokaryote could be deduced from the sequence of its proteome. Finally, we aimed at finding the precise differences between proteome sequences of prokaryotes from different environments.
Rajesh Ramaswamy Partial-propensity simulation algorithms for stochastic chemical kinetics and the role of fluctuations in mesoscopic reaction systems
Ph.D. Thesis, ETH Zurich, Department of Computer Science, Zurich, Switzerland (2011) PDF
Rajesh Ramaswamy, Ivo F. Sbalzarini, Nélido González-Segredo Noise-induced modulation of the relaxation kinetics around a non-equilibrium steady state of non-linear chemical reaction networks PLoS ONE, 6(1) Art. No. e16045 (2011)
PDF
DOI
Stochastic effects from correlated noise non-trivially modulate the kinetics of non-linear chemical reaction networks. This is especially important in systems where reactions are confined to small volumes and reactants are delivered in bursts. We characterise how the two noise sources confinement and burst modulate the relaxation kinetics of a non-linear reaction network around a non-equilibrium steady state. We find that the lifetimes of species change with burst input and confinement. Confinement increases the lifetimes of all species that are involved in any non-linear reaction as a reactant. Burst monotonically increases or decreases lifetimes. Competition between burst-induced and confinement-induced modulation may hence lead to a non-monotonic modulation. We quantify lifetime as the integral of the time autocorrelation function (ACF) of concentration fluctuations around a non-equilibrium steady state of the reaction network. Furthermore, we look at the first and second derivatives of the ACF, each of which is affected in opposite ways by burst and confinement. This allows discriminating between these two noise sources. We analytically derive the ACF from the linear Fokker-Planck approximation of the chemical master equation in order to establish a baseline for the burst-induced modulation at low confinement. Effects of higher confinement are then studied using a partial-propensity stochastic simulation algorithm. The results presented here may help understand the mechanisms that deviate stochastic kinetics from its deterministic counterpart. In addition, they may be instrumental when using fluorescence-lifetime imaging microscopy (FLIM) or fluorescence-correlation spectroscopy (FCS) to measure confinement and burst in systems with known reaction rates, or, alternatively, to correct for the effects of confinement and burst when experimentally measuring reaction rates.
Rajesh Ramaswamy, Ivo F. Sbalzarini Intrinsic noise alters the frequency spectrum of mesoscopic oscillatory chemical reaction systems Sci Rep, 1 Art. No. 154 (2011)
PDF
DOI
Mesoscopic oscillatory reaction systems, for example in cell biology, can exhibit stochastic oscillations in the form of cyclic random walks even if the corresponding macroscopic system does not oscillate. We study how the intrinsic noise from molecular discreteness influences the frequency spectrum of mesoscopic oscillators using as a model system a cascade of coupled Brusselators away from the Hopf bifurcation. The results show that the spectrum of an oscillator depends on the level of noise. In particular, the peak frequency of the oscillator is reduced by increasing noise, and the bandwidth increased. Along a cascade of coupled oscillators, the peak frequency is further reduced with every stage and also the bandwidth is reduced. These effects can help understand the role of noise in chemical oscillators and provide fingerprints for more reliable parameter identification and volume measurement from experimental spectra.
Christian L. Müller, Ivo F. Sbalzarini Global characterization of the CEC 2005 fitness landscapes using fitness-distance analysis
In: Proc. EvoStar
(2011) Leture Notes in Computer Science, New York, Springer (2011), 294-303
PDF
Christian L. Müller, Ivo F. Sbalzarini Gaussian Adaptation for robust design centering
In: Proc. EuroGen
(2011), Italy, CIRA (2011), 736-742
PDF
Birte Schrader Discretization-Corrected PSE Operators for Adaptive Multiresolution Particle Methods
Ph.D. Thesis, ETH Zurich, Department of Computer Science, Zurich, Switzerland (2011) PDF
Christian L. Müller Continuous black-box optimization in linearly constrained domains using efficient Gibbs sampling
In: Proc. EVOLVE
(2011), Luxembourg, EVOLVE (2011), 1-4
PDF
Rajesh Ramaswamy, Ivo F. Sbalzarini A partial-propensity formulation of the stochastic simulation algorithm for chemical reaction networks with delays J Chem Phys, 134(1) Art. No. 014106 (2011)
PDF
DOI
Several real-world systems, such as gene expression networks in biological cells, contain coupled chemical reactions with a time delay between reaction initiation and completion. The non-Markovian kinetics of such reaction networks can be exactly simulated using the delay stochastic simulation algorithm (dSSA). The computational cost of dSSA scales with the total number of reactions in the network. We reduce this cost to scale at most with the smaller number of species by using the concept of partial reaction propensities. The resulting delay partial-propensity direct method (dPDM) is an exact dSSA formulation for well-stirred systems of coupled chemical reactions with delays. We detail dPDM and present a theoretical analysis of its computational cost. Furthermore, we demonstrate the implications of the theoretical cost analysis in two prototypical benchmark applications. The dPDM formulation is shown to be particularly efficient for strongly coupled reaction networks, where the number of reactions is much larger than the number of species.
Ömer Demirel, Birte Schrader, Ivo F. Sbalzarini A parallel particle method for solving the EEG source localization forward problem
In: Proc. 6th Intl. Symp. Health Informatics and Bioinformatics (HIBIT)
(2011), Piscataway, N.J., IEEE (2011), 154-158
PDF
Gregory Paul, Janick Cardinale, Ivo F. Sbalzarini An alternating split Bregman algorithm for multi-region segmentation
In: Proc. 45th IEEE Asilomar Conf. Signals, Systems, and Computers
(2011), Piscataway, N.J., IEEE (2011), 426-430
PDF
Christian L. Müller, Ivo F. Sbalzarini A conjecture about an upper bound of the RMSD between linear chains
In: Proc. EuroCG
(2011), Brussels, EuroCG (2011), 31-34
PDF
2010
M. Bergdorf, Ivo F. Sbalzarini, Petros Koumoutsakos A Lagrangian particle method for reaction-diffusion systems on deforming surfaces. J Math Biol, 61(5) 649-663 (2010)
PDF
DOI
Reaction-diffusion processes on complex deforming surfaces are fundamental to a number of biological processes ranging from embryonic development to cancer tumor growth and angiogenesis. The simulation of these processes using continuum reaction-diffusion models requires computational methods capable of accurately tracking the geometric deformations and discretizing on them the governing equations. We employ a Lagrangian level-set formulation to capture the deformation of the geometry and use an embedding formulation and an adaptive particle method to discretize both the level-set equations and the corresponding reaction-diffusion. We validate the proposed method and discuss its advantages and drawbacks through simulations of reaction-diffusion equations on complex and deforming geometries.
Omar Awile, Ömer Demirel, Ivo F. Sbalzarini Toward an Object-Oriented Core of the PPM Library
In: Numerical Analysis and Applied Mathematics : International Conference on Numerical Analysis and Applied Mathematics ; Rhodes, Greece, 19 - 25 September 2010 ; ICNAAM 2010
(2010)(Eds.) Theodore E. Simos AIP conference proceedings ; 1281, Melville, N.Y., AIP (2010), 1313-1316
PDF
Omar Awile, Anita Krisko, Ivo F. Sbalzarini, Bojan Zagrovic Intrinsically disordered regions may lower the hydration free energy in proteins: a case study of nudix hydrolase in the bacterium Deinococcus radiodurans. PLoS Comput Biol, 6(7) 1000854-1000854 (2010)
Open AccessPDF
DOI
The proteome of the radiation- and desiccation-resistant bacterium D. radiodurans features a group of proteins that contain significant intrinsically disordered regions that are not present in non-extremophile homologues. Interestingly, this group includes a number of housekeeping and repair proteins such as DNA polymerase III, nudix hydrolase and rotamase. Here, we focus on a member of the nudix hydrolase family from D. radiodurans possessing low-complexity N- and C-terminal tails, which exhibit sequence signatures of intrinsic disorder and have unknown function. The enzyme catalyzes the hydrolysis of oxidatively damaged and mutagenic nucleotides, and it is thought to play an important role in D. radiodurans during the recovery phase after exposure to ionizing radiation or desiccation. We use molecular dynamics simulations to study the dynamics of the protein, and study its hydration free energy using the GB/SA formalism. We show that the presence of disordered tails significantly decreases the hydration free energy of the whole protein. We hypothesize that the tails increase the chances of the protein to be located in the remaining water patches in the desiccated cell, where it is protected from the desiccation effects and can function normally. We extrapolate this to other intrinsically disordered regions in proteins, and propose a novel function for them: intrinsically disordered regions increase the "surface-properties" of the folded domains they are attached to, making them on the whole more hydrophilic and potentially influencing, in this way, their localization and cellular activity.
Christian L. Müller, Ivo F. Sbalzarini Gaussian adaptation revisited — an entropic view on covariance matrix adaptation
In: Proc. EvoStar
(2010) Lecture Notes in Computer Science, Berlin;Heidelberg, Springer (2010), 432-441
PDF
Christian L. Müller, Ivo F. Sbalzarini Gaussian Adaptation as a unifying framework for continuous black-box optimization and adaptive Monte Carlo sampling
In: Proc. IEEE Congress on Evolutionary Computation (CEC)
(2010), Piscataway, N.J., IEEE (2010), 2594-2601
PDF
Rajesh Ramaswamy, Ivo F. Sbalzarini Fast exact stochastic simulation algorithms using partial propensities
In: Proc. ICNAAM
(2010), Melville, N.Y., AIP (2010), 1338-1341
PDF
Birte Schrader, Sylvain Reboux, Ivo F. Sbalzarini Discretization correction of general integral PSE Operators for particle methods J Comput Phys, 229 4159-4182 (2010)
PDF
Birte Schrader, Sylvain Reboux, Ivo F. Sbalzarini Discretization-Corrected PSE Operators for Particle Methods
In: Proc. 8th Euromech Fluid Mechanics Conference (EFMC8)
(2010), EuroMech EFMC (2010), 1-1
PDF
Jo A. Helmuth Computational Methods for Analyzing and Simulating Intra-Cellular Transport Processes
Ph.D. Thesis, ETH Zurich, Department of Computer Science, Zurich, Switzerland (2010) PDF
Christian L. Müller Black-box Landscapes: Characterization, Optimization, Sampling, and Application to Geometric Configuration Problems
Ph.D. Thesis, ETH Zurich, Department of Computer Science, Zurich, Switzerland (2010) PDF
Jo A. Helmuth, Gregory Paul, Ivo F. Sbalzarini Beyond co-localization: inferring spatial interactions between sub-cellular structures from microscopy images BMC Bioinformatics, 11 Art. No. 372 (2010)
PDF
DOI
Sub-cellular structures interact in numerous direct and indirect ways in order to fulfill cellular functions. While direct molecular interactions crucially depend on spatial proximity, other interactions typically result in spatial correlations between the interacting structures. Such correlations are the target of microscopy-based co-localization analysis, which can provide hints of potential interactions. Two complementary approaches to co-localization analysis can be distinguished: intensity correlation methods capitalize on pattern discovery, whereas object-based methods emphasize detection power.
Rajesh Ramaswamy, Ivo F. Sbalzarini A partial-propensity variant of the composition-rejection stochastic simulation algorithm for chemical reaction networks J Chem Phys, 132(4) Art. No. 044102 (2010)
PDF
DOI
We present the partial-propensity stochastic simulation algorithm with composition-rejection sampling (PSSA-CR). It is an exact formulation of the stochastic simulation algorithm (SSA) for well-stirred systems of coupled chemical reactions. The new formulation is a partial-propensity variant [R. Ramaswamy, N. Gonzalez-Segredo, and I. F. Sbalzarini, J. Chem. Phys. 130, 244104 (2009)] of the composition- rejection SSA [A. Slepoy, A. P. Thompson, and S. J. Plimpton, J. Chem. Phys. 128, 205101 (2008)]. The computational cost of this new formulation is bounded by a constant for weakly coupled reaction networks, and it increases at most linearly with the number of chemical species for strongly coupled reaction networks. PSSA-CR thus combines the advantages of partial-propensity methods and the composition-rejection SSA, providing favorable scaling of the computational cost for all classes of reaction networks.
Anton A. Polyansky, Rajesh Ramaswamy, Pavel E. Volynsky, Ivo F. Sbalzarini, Siewert J. Marrink, Roman G. Efremov Antimicrobial peptides induce growth of phosphatidylglycerol domains in a model bacterial membrane J Phys Chem Lett, 1 3108-3111 (2010)
PDF
DOI
Ivo F. Sbalzarini Abstractions and middleware for petascale computing and beyond Intl J Dist Sys & Technol, 1(2) 40-56 (2010)
PDF
2009
Jo A. Helmuth#, C J Burckhardt, U F Greber, Ivo F. Sbalzarini# Shape reconstruction of subcellular structures from live cell fluorescence microscopy images J Struct Biol, 167(1) 1-10 (2009)
PDF
DOI
Live imaging of subcellular structures is indispensible to advance our understanding of cellular processes. The blurred digital images acquired in light microscopy are, however, complex to analyze, and identification and reconstruction of subcellular structures from such images remains a major challenge. We present a novel, model-based image analysis algorithm to reconstruct outlines of subcellular structures using a sub-pixel representation. The algorithm explicitly accounts for the optical properties of the microscope. We validate the reconstruction performance on synthetic data and apply the new method to fluorescence microscopy images of endosomes identified by the GTPase EGFP-Rab5. The benefits of the new algorithm are outlined by comparison to standard techniques. We demonstrate that the new algorithm leads to better discrimination between different endosomal virus entry pathways and to more robust, accurate, and self-consistent quantification of endosome shape features. This allows establishing a set of features that quantify endosome morphology and robustly capture the dynamics of endosome fusion.
Christian L. Müller, Ivo F. Sbalzarini, Wilfred F van Gunsteren, Bojan Zagrović, Philippe H Hünenberger In the eye of the beholder: Inhomogeneous distribution of high-resolution shapes within the random-walk ensemble J Chem Phys, 130(21) Art. No. 214904 (2009)
PDF
DOI
The concept of high-resolution shapes (also referred to as folds or states, depending on the context) of a polymer chain plays a central role in polymer science, structural biology, bioinformatics, and biopolymer dynamics. However, although the idea of shape is intuitively very useful, there is no unambiguous mathematical definition for this concept. In the present work, the distributions of high-resolution shapes within the ideal random-walk ensembles with N=3,...,6 beads (or up to N=10 for some properties) are investigated using a systematic (grid-based) approach based on a simple working definition of shapes relying on the root-mean-square atomic positional deviation as a metric (i.e., to define the distance between pairs of structures) and a single cutoff criterion for the shape assignment. Although the random-walk ensemble appears to represent the paramount of homogeneity and randomness, this analysis reveals that the distribution of shapes within this ensemble, i.e., in the total absence of interatomic interactions characteristic of a specific polymer (beyond the generic connectivity constraint), is significantly inhomogeneous. In particular, a specific (densest) shape occurs with a local probability that is 1.28, 1.79, 2.94, and 10.05 times (N=3,...,6) higher than the corresponding average over all possible shapes (these results can tentatively be extrapolated to a factor as large as about 10(28) for N=100). The qualitative results of this analysis lead to a few rather counterintuitive suggestions, namely, that, e.g., (i) a fold classification analysis applied to the random-walk ensemble would lead to the identification of random-walk "folds;" (ii) a clustering analysis applied to the random-walk ensemble would also lead to the identification random-walk "states" and associated relative free energies; and (iii) a random-walk ensemble of polymer chains could lead to well-defined diffraction patterns in hypothetical fiber or crystal diffraction experiments. The inhomogeneous nature of the shape probability distribution identified here for random walks may represent a significant underlying baseline effect in the analysis of real polymer chain ensembles (i.e., in the presence of specific interatomic interactions). As a consequence, a part of what is called a polymer shape may actually reside just "in the eye of the beholder" rather than in the nature of the interactions between the constituting atoms, and the corresponding observation-related bias should be taken into account when drawing conclusions from shape analyses as applied to real structural ensembles.
Rajesh Ramaswamy, Nélido González-Segredo, Ivo F. Sbalzarini A new class of highly efficient exact stochastic simulation algorithms for chemical reaction networks J Chem Phys, 130(24) Art. No. 244104 (2009)
PDF
DOI
We introduce an alternative formulation of the exact stochastic simulation algorithm (SSA) for sampling trajectories of the chemical master equation for a well-stirred system of coupled chemical reactions. Our formulation is based on factored-out, partial reaction propensities. This novel exact SSA, called the partial-propensity direct method (PDM), is highly efficient and has a computational cost that scales at most linearly with the number of chemical species, irrespective of the degree of coupling of the reaction network. In addition, we propose a sorting variant, SPDM, which is especially efficient for multiscale reaction networks.
Harvey A Zambrano, Jens H. Walther, Petros Koumoutsakos, Ivo F. Sbalzarini Thermophoretic motion of water nanodroplets confined inside carbon nanotubes. Nano Lett, 9(1) 66-71 (2009)
PDF
DOI
We study the thermophoretic motion of water nanodroplets confined inside carbon nanotubes using molecular dynamics simulations. We find that the nanodroplets move in the direction opposite the imposed thermal gradient with a terminal velocity that is linearly proportional to the gradient. The translational motion is associated with a solid body rotation of the water nanodroplet coinciding with the helical symmetry of the carbon nanotube. The thermal diffusion displays a weak dependence on the wetting of the water-carbon nanotube interface. We introduce the use of the moment scaling spectrum (MSS) in order to determine the characteristics of the motion of the nanoparticles inside the carbon nanotube. The MSS indicates that affinity of the nanodroplet with the walls of the carbon nanotubes is important for the isothermal diffusion and hence for the Soret coefficient of the system.
Ivo F. Sbalzarini Spatiotemporal Modeling and Simulation in Biology
In: Bioinformatics., Hackensack, USA, World Scientific (2009), 381-432 Ch. 14 PDF
Christian L. Müller, Benedikt Baumgartner, Georg Ofenbeck, Birte Schrader, Ivo F. Sbalzarini pCMALib: a parallel FORTRAN 90 library for the evolution strategy with covariance matrix adaptation
In: Proc. ACM Genetic and Evolutionary Computation Conference (GECCO’09)
(2009), New York, ACM (2009), 1-8
PDF
Christian L. Müller, Benedikt Baumgartner, Ivo F. Sbalzarini Particle Swarm CMA Evolution Strategy for the Optimization of Multi-Funnel Landscapes
In: IEEE Congress on Evolutionary Computation : CEC 2009 ; Trondheim, Norway, 18 - 21 May 2009
(2009), Piscataway, N.J., IEEE (2009), 2685-2692
PDF
Jens H. Walther, Ivo F. Sbalzarini Large-scale parallel discrete element simulations of granular flows Eng Comput, 26(6) 688-697 (2009)
DOI
Jo A. Helmuth, Ivo F. Sbalzarini Deconvolving Active Contours for Fluorescence Microscopy Images
In: Advances in Visual Computing : 5th International Symposium Visual Computing ISVC 2009, Las Vegas, NV; USA, Nov. 30-Dec. 02, 2009 ; Poceedings, Part 1
(2009)(Eds.) George Bebis Lecture Notes in Computer Science ; 5875, Berlin;Heidelberg, Springer (2009), 544-553
PDF
Jo A. Helmuth, Ivo F. Sbalzarini Deconvolving active contours for fluorescence microscopy images
In: Proc. Intl. Symp. Visual Computing (ISVC)
(2009) Lecture Notes in Computer Science, Berlin;Heidelberg, Springer (2009), 544-553
PDF
Janick Cardinale, Alexander Rauch, Yves Barral, Gabor Szekely, Ivo F. Sbalzarini Bayesian image analysis with on-line confidence estimates and its application to microtubule tracking
In: In Proc. IEEE Intl. Symposium Biomedical Imaging (ISBI)
(2009), Piscataway, N.J., IEEE (2009), 1091-1094
PDF
Christian L. Müller, Ivo F. Sbalzarini A tunable real-world multi-funnel benchmark problem for evolutionary optimization (and why parallel island models might remedy the failure of CMA-ES on it)
In: Proc. Intl. Joint Conf. Computational Intelligence (IJCCI)
(2009), INSTICC Press (2009), 248-253
PDF
Ivo F. Sbalzarini Analysis, Modeling & Simulation of Diffusion Processes in Cell Biology
Heidelberg, Germany, VDM Verlag (2009), 388 S.
2007
Jo A. Helmuth, C J Burckhardt, Petros Koumoutsakos, U F Greber, Ivo F. Sbalzarini A novel supervised trajectory segmentation algorithm identifies distinct types of human adenovirus motion in host cells J Struct Biol, 159(3) 347-358 (2007)
PDF
DOI
Biological trajectories can be characterized by transient patterns that may provide insight into the interactions of the moving object with its immediate environment. The accurate and automated identification of trajectory motifs is important for the understanding of the underlying mechanisms. In this work, we develop a novel trajectory segmentation algorithm based on supervised support vector classification. The algorithm is validated on synthetic data and applied to the identification of trajectory fingerprints of fluorescently tagged human adenovirus particles in live cells. In virus trajectories on the cell surface, periods of confined motion, slow drift, and fast drift are efficiently detected. Additionally, directed motion is found for viruses in the cytoplasm. The algorithm enables the linking of microscopic observations to molecular phenomena that are critical in many biological processes, including infectious pathogen entry and signal transduction.
Jens H. Walther#, Ivo F. Sbalzarini# Large-scale parallel discrete element simulations of granular flow
In: Proceedings of the International Conference on Discrete Element Modelling (DEM07)
(2007), Amsterdam, Netherlands, Academic Press (2007), 1-8
PDF
Ivo F. Sbalzarini Informatik und Biologie – Eine Symbiose ermöglicht neue Entdeckungen Vierteljahresschrift der Naturforschenden Gesellschaft in Zurich, 152(3) 63-70 (2007)
PDF
2006
Ivo F. Sbalzarini, Arnold Hayer, Ari Helenius, Petros Koumoutsakos Simulations of (an)isotropic diffusion on curved biological surfaces. Biophys J, 90(3) 878-885 (2006)
PDF
DOI
We present a computational particle method for the simulation of isotropic and anisotropic diffusion on curved biological surfaces that have been reconstructed from image data. The method is capable of handling surfaces of high curvature and complex shape, which are often encountered in biology. The method is validated on simple benchmark problems and is shown to be second-order accurate in space and time and of high parallel efficiency. It is applied to simulations of diffusion on the membrane of endoplasmic reticula (ER) in live cells. Diffusion simulations are conducted on geometries reconstructed from real ER samples and are compared to fluorescence recovery after photobleaching experiments in the same ER samples using the transmembrane protein tsO45-VSV-G, C-terminally tagged with green fluorescent protein. Such comparisons allow derivation of geometry-corrected molecular diffusion constants for membrane components from fluorescence recovery after photobleaching data. The results of the simulations indicate that the diffusion behavior of molecules in the ER membrane differs significantly from the volumetric diffusion of soluble molecules in the lumen of the same ER. The apparent speed of recovery differs by a factor of approximately 4, even when the molecular diffusion constants of the two molecules are identical. In addition, the specific shape of the membrane affects the recovery half-time, which is found to vary by a factor of approximately 2 in different ER samples.
Ivo F. Sbalzarini, J. Walther, M. Bergdorf, Simone E. Hieber, Evangelos M. Kotsalis, Petros Koumoutsakos PPM – A highly efficient parallel particle–mesh library for the simulation of continuum systems J Comput Phys, 215 566-588 (2006)
PDF
Ivo F. Sbalzarini, Jens H. Walther, Bettina Polasek, Philippe Chatelain, M. Bergdorf, Simone E. Hieber, Evangelos M. Kotsalis, Petros Koumoutsakos A Software Framework for Portable Parallelization of Particle-Mesh Simulations
In: Proc. EuroPar
(2006)(Eds.) Wolfgang E Nagel Lecture Notes in Computer Science, New York, Springer (2006), 730-739
PDF
Ivo F. Sbalzarini Analysis, Modeling, and Simulation of Diffusion Processes in Cell Biology
Ph.D. Thesis, ETH Zurich, Department of Computer Science, Zurich, Switzerland (2006) PDF
2005
Helge Ewers, Alicia E. Smith, Ivo F. Sbalzarini, Hauke Lilie, Petros Koumoutsakos, Ari Helenius Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes Proc Natl Acad Sci U.S.A., 102(42) 15110-15115 (2005)
PDF
DOI
The lateral mobility of individual murine polyoma virus-like particles (VLPs) bound to live cells and artificial lipid bilayers was studied by single fluorescent particle tracking using total internal reflection fluorescence microscopy. The particle trajectories were analyzed in terms of diffusion rates and modes of motion as described by the moment scaling spectrum. Although VLPs bound to their ganglioside receptor in lipid bilayers exhibited only free diffusion, analysis of trajectories on live 3T6 mouse fibroblasts revealed three distinct modes of mobility: rapid random motion, confined movement in small zones (30-60 nm in diameter), and confined movement in zones with a slow drift. After binding to the cell surface, particles typically underwent free diffusion for 5-10 s, and then they were confined in an actin filament-dependent manner without involvement of clathrin-coated pits or caveolae. Depletion of cholesterol dramatically reduced mobility of VLPs independently of actin, whereas inhibition of tyrosine kinases had no effect on confinement. The results suggested that clustering of ganglioside molecules by the multivalent VLPs induced transmembrane coupling that led to confinement of the virus/receptor complex by cortical actin filaments.
Ivo F. Sbalzarini, Anna Mezzacasa, Ari Helenius, Petros Koumoutsakos Effects of organelle shape on fluorescence recovery after photobleaching Biophys J, 89(3) 1482-1492 (2005)
PDF
DOI
The determination of diffusion coefficients from fluorescence recovery data is often complicated by geometric constraints imposed by the complex shapes of intracellular compartments. To address this issue, diffusion of proteins in the lumen of the endoplasmic reticulum (ER) is studied using cell biological and computational methods. Fluorescence recovery after photobleaching (FRAP) experiments are performed in tissue culture cells expressing GFP-KDEL, a soluble, fluorescent protein, in the ER lumen. The three-dimensional (3D) shape of the ER is determined by confocal microscopy and computationally reconstructed. Within these ER geometries diffusion of solutes is simulated using the method of particle strength exchange. The simulations are compared to experimental FRAP curves of GFP-KDEL in the same ER region. Comparisons of simulations in the 3D ER shapes to simulations in open 3D space show that the constraints imposed by the spatial confinement result in two- to fourfold underestimation of the molecular diffusion constant in the ER if the geometry is not taken into account. Using the same molecular diffusion constant in different simulations, the observed speed of fluorescence recovery varies by a factor of 2.5, depending on the particular ER geometry and the location of the bleached area. Organelle shape considerably influences diffusive transport and must be taken into account when relating experimental photobleaching data to molecular diffusion coefficients. This novel methodology combines experimental FRAP curves with high accuracy computer simulations of diffusion in the same ER geometry to determine the molecular diffusion constant of the solute in the particular ER lumen.
Ivo F. Sbalzarini, Petros Koumoutsakos Feature point tracking and trajectory analysis for video imaging in cell biology J Struct Biol, 151(2) 182-195 (2005)
PDF
DOI
This paper presents a computationally efficient, two-dimensional, feature point tracking algorithm for the automated detection and quantitative analysis of particle trajectories as recorded by video imaging in cell biology. The tracking process requires no a priori mathematical modeling of the motion, it is self-initializing, it discriminates spurious detections, and it can handle temporary occlusion as well as particle appearance and disappearance from the image region. The efficiency of the algorithm is validated on synthetic video data where it is compared to existing methods and its accuracy and precision are assessed for a wide range of signal-to-noise ratios. The algorithm is well suited for video imaging in cell biology relying on low-intensity fluorescence microscopy. Its applicability is demonstrated in three case studies involving transport of low-density lipoproteins in endosomes, motion of fluorescently labeled Adenovirus-2 particles along microtubules, and tracking of quantum dots on the plasma membrane of live cells. The present automated tracking process enables the quantification of dispersive processes in cell biology using techniques such as moment scaling spectra.
Cosima Luedeke, Stéphanie Buvelot Frei, Ivo F. Sbalzarini, Heinz Schwarz, Anne Spang, Yves Barral Septin-dependent compartmentalization of the endoplasmic reticulum during yeast polarized growth. J Cell Biol, 169(6) 897-908 (2005)
PDF
DOI
Polarized cells frequently use diffusion barriers to separate plasma membrane domains. It is unknown whether diffusion barriers also compartmentalize intracellular organelles. We used photobleaching techniques to characterize protein diffusion in the yeast endoplasmic reticulum (ER). Although a soluble protein diffused rapidly throughout the ER lumen, diffusion of ER membrane proteins was restricted at the bud neck. Ultrastructural studies and fluorescence microscopy revealed the presence of a ring of smooth ER at the bud neck. This ER domain and the restriction of diffusion for ER membrane proteins through the bud neck depended on septin function. The membrane-associated protein Bud6 localized to the bud neck in a septin-dependent manner and was required to restrict the diffusion of ER membrane proteins. Our results indicate that Bud6 acts downstream of septins to assemble a fence in the ER membrane at the bud neck. Thus, in polarized yeast cells, diffusion barriers compartmentalize the ER and the plasma membrane along parallel lines.
Ivo F. Sbalzarini, Helge Ewers, Alicia E. Smith, Ari Helenius, Petros Koumoutsakos Heimtückische Viren auf lebenden Zellen ETH Bulletin, 298 48-50 (2005)
PDF
2004
Ivo F. Sbalzarini, Anna Mezzacasa, Ari Helenius, Petros Koumoutsakos Large-Scale Simulations of Diffusion in Cell Biology ERCIM News, 59 69-70 (2004)
PDF
2002
Ivo F. Sbalzarini Diffusion in the Endoplasmic Reticulum
Diploma Thesis,ETH Zurich, Department of Computer Science, Zurich, Switzerland (2002) PDF
2001
Ivo F. Sbalzarini, Sibylle D. Müller, Petros Koumoutsakos Microchannel Optimization Using Multiobjective Evolution Strategies
(2001) Lecture Notes in Computer Science, New York, Springer (2001), 516-530
PDF
Sibylle D. Müller, Ivo F. Sbalzarini, Jens H. Walther, Petros Koumoutsakos Evolution Strategies for the Optimization of Microdevices
In: Proceedings of the Congress on Evolutionary Computation (CEC 2001)
(2001), Piscataway, N.J., IEEE (2001), 302-309
PDF
Ivo F. Sbalzarini, Sibylle D. Müller, Petros Koumoutsakos#, Georges-Henri Cottet# Evolution Strategies for Computational and Experimental Fluid Dynamics Applications
In: Proceedings of the Genetic and Evolutionary Computation Conference (GECCO 2001)
(2001), New York, ACM (2001), 1-8
PDF