seeks to understand protein sequence space in the light of evolution and erroneous protein production with focus on intrinsically disordered and condensate-forming proteins. We complement the increasing amount of high-throughput data with rigorous computational studies, and provide hypotheses for experimental validation.

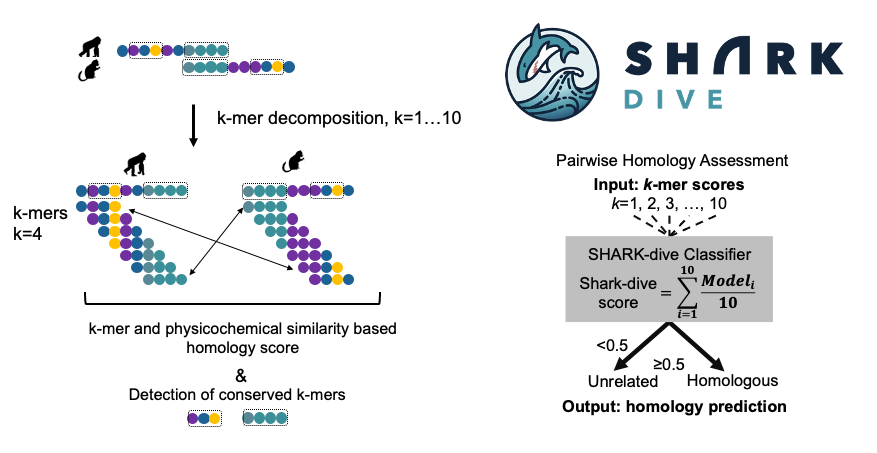
Increasing insights into how sequence motifs in intrinsically disordered regions (IDRs) provide functions underscore the need for systematic motif detection. Contrary to structured regions where motifs can be readily identified from sequence alignments, the rapid evolution of IDRs limits the usage of alignment-based tools in reliably detecting motifs within.
Here, we developed SHARK-capture (10.1002/pro.70091), an alignment-free motif detection tool designed for difficult-to-align regions. SHARK-capture innovates on word-based methods by flexibly incorporating amino acid physicochemistry to assess motif similarity without requiring rigid definitions of equivalency groups and offers consistently strong performance in a systematic benchmark, with superior residue-level performance.
Chi Fung Willis Chow, Swantje Lenz, Maxim Scheremetjew, Soumyadeep Ghosh, Doris Richter, Ceciel Jegers, Alexander von Appen, Simon Alberti, Agnes Toth-Petroczy. SHARK-capture identifies functional motifs in intrinsically disordered protein regions. Protein Sci. 2025 Apr;34(4):e70091. doi: 10.1002/pro.70091. PMID: 40100159; PMCID: PMC11917139.
.
Most of our understanding about the molecular history of the cell is biased towards well-conserved ordered protein domains. Intrinsically disordered protein regions (IDRs) are largely unexplored due to their low sequence complexity and low conservation. Yet they have pivotal roles in the cell including the formation of biomolecular condensates.
We have designed an alignment-free algorithm to assess homology between un-ulignable IDR sequences (SHARK-dive, 10.1073/pnas.2401622121) and developed a method to identify conserved motifs within a set of homologous IDR sequences (SHARK-capture). Based on evolutionary information, we can classify IDR families using our new tools, and propose functional sites that we are validating experimentally.
Chi Fung Willis Chow, Soumyadeep Ghosh, Anna Hadarovich, and Agnes Toth-Petroczy. SHARK enables sensitive detection of evolutionary homologs and functional analogs in unalignable and disordered sequences. Proc Natl Acad Sci U S A. 2024 Oct 15;121(42):e2401622121. doi: 10.1073/pnas.2401622121. Epub 2024 Oct 9. PMID: 39383002.
To integrate interdisciplinary scientific knowledge about the function and composition of biomolecular condensates, we developed the CrowDsourcing COndensate Database and Encyclopedia (CD-CODE.org). CD-CODE is a community-editable platform, which includes a database of biomolecular condensates based on the literature, an encyclopedia of relevant scientific terms, and a crowdsourcing web application.
Ksenia Kuznetsova, Maxim Scheremetjew, Jialin Yin, HongKee Moon, Diego A Vargas, Anna Hadarovich, Natasha Lewis, Carsten Hoege, Chi Fung Willis Chow, David Kuster, Jik Nijssen, Alberto Hernandez-Armendariz, Jonathan C Savage, Yu Wei, Silja Zedlitz, Hari Raj Singh, Soumyadeep Ghosh, Allysa P Kemraj, Lena Hersemann, Anthony A Hyman, Diana M Mitrea, Agnes Toth-Petroczy. CD-CODE 2.0: an enhanced condensate knowledgebase integrating pathobiology, condensate modulating drugs, and host–pathogen interactions. Nucleic Acids Research, 2025, gkaf1104. Open access
Behind the paper blog post: Condensing information on condensates
MPI-CBG News: CD-CODE – Database and Encyclopedia for membraneless liquid droplets
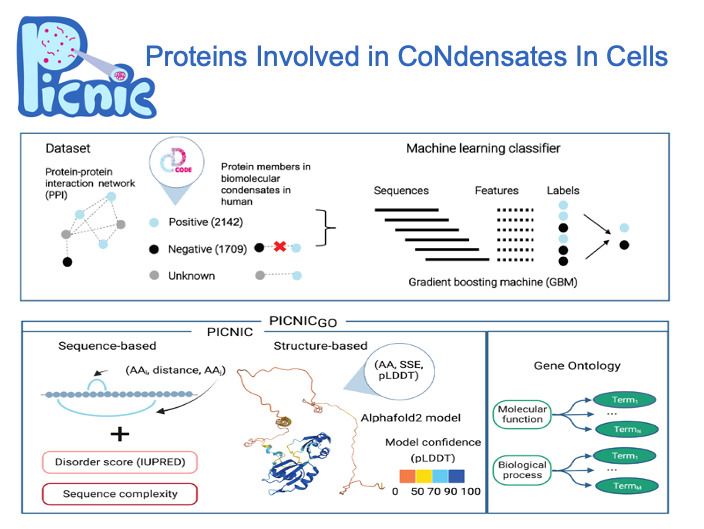
To enable systematic detection of condensate proteins, we developed an algorithm to recognize proteins involved in in vivo biomolecular condensates regardless of the mechanism of condensate formation. Using a curated dataset of CD-CODE, we trained a machine learning classifier based on sequence- and structure-based features to predict if a protein is part of a condensate. Our model, PICNIC (Proteins Involved in CoNdensates In Cells) enables systematic detection of condensate proteins across organisms.
We show that protein sequence and structure encode the condensate forming function. PICNIC accurately predicts which proteins are involved in condensates, as 18 out of 24 proteins tested experimentally indeed formed high-confidence in cellulo condensates. We infer that ~40% of the proteomes of organisms across the tree of life form condensates with no apparent correlation with disorder content and organismal complexity.
Precomputed scores for 14 organisms are available at picnic.cd-code.org.
Hadarovich A, Singh HR, Ghosh S, Scheremetjew M, Rostam N, Hyman AA, Toth-Petroczy A. PICNIC accurately predicts condensate-forming proteins regardless of their structural disorder across organisms. Nat Commun. 2024 Dec 11;15(1):10668. doi: 10.1038/s41467-024-55089-x. PMID: 39663388.
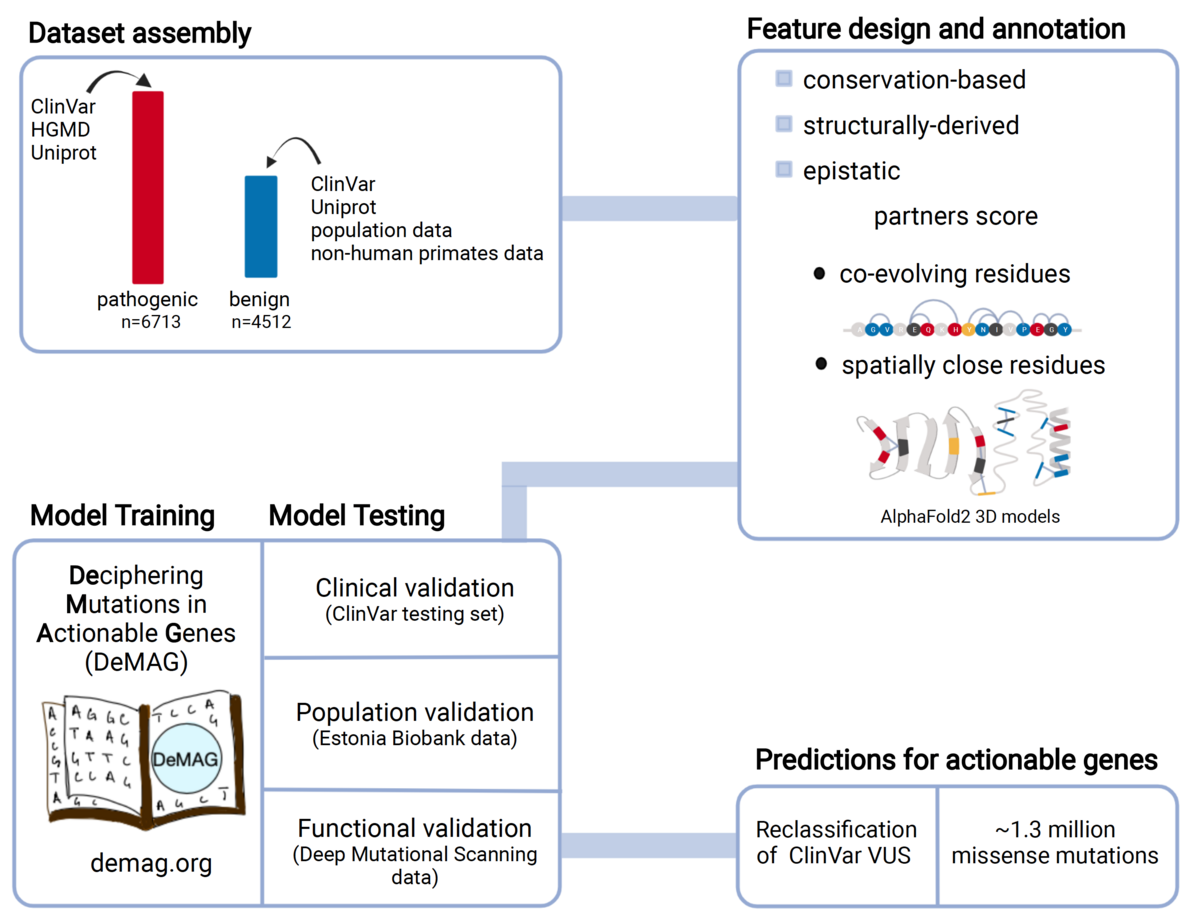
Assessing the phenotypic effects of single nucleotide variants remains a challenge even in genes with well-established clinical impact. We have developed DeMAG (Deciphering Mutations in Actionable Genes, demag.org) that reaches unprecedented accuracy in the classification of missense variants in clinically actionable genes. Our approach makes use of protein 3D structures and epistatic residue interactions to derive new predictive features, as well as abundant clinical diagnostic data in these genes to improve performance.
Luppino, Federica, Ivan A. Adzhubei, Christopher A. Cassa, and Agnes Toth-Petroczy. 2023. “DeMAG Predicts the Effects of Variants in Clinically Actionable Genes by Integrating Structural and Evolutionary Epistatic Features.” Nature Communications 2023, 14 (1): 1–14. Open Access
MPI-CBG News article: New tool facilitates clinical interpretation of genetic information.
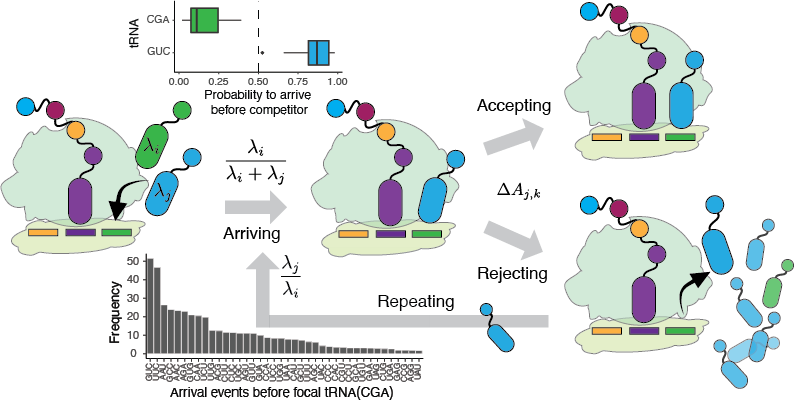
Transcriptional and translational errors generate diverse sequences, most dysfunctional, some harboring novel functions (Romero et. al, Prot Sci. 2022).
To detect and quantify phenotypic mutations proteome-wide, we combine theoretical modeling, machine learning and experiments. We developed a theoretical model and mass spectrometry pipeline to study amino acid misincoroprations at proteome scale (Landerer et. al, Mol Bio Evol 2024).
Using a library of reporters for STOP codon miscoding in E. coli, we found that under stress conditions, stop codon miscoding may occur with a rate as high as 80%, depending on the nucleotide context (Romero et. al, Nat. Commu. 2024). Further, our analysis of selected reporters by mass spectrometry and RNA-seq showed that not only translation but also transcription errors contribute to stop codon miscoding. The RNA polymerase is more likely to misincorporate a nucleotide at premature stop codons.
We are also developing algorithms for predicting frameshifts by using proteomics data. We aim at discovering novel frameshift and STOP codon read-through variants, and we will explore the evolutionary potential of slippage sites and test whether they could lead to promiscuous protein functions.