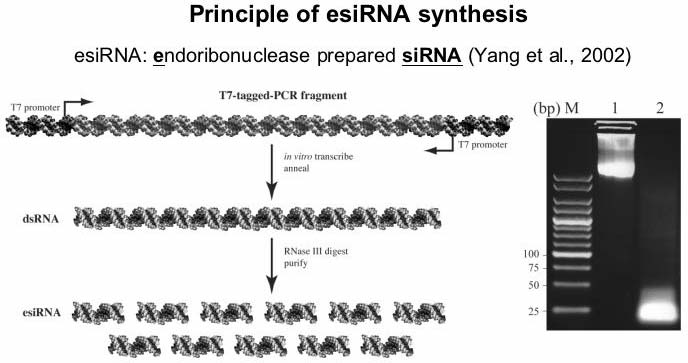
Schematic presentation of esiRNA production. Steps carried out to obtain a pool of functional siRNAs are illustrated. An agarose gel before (lane 1) and after (lane 2) RNaseIII digestion is shown.
Here you find a general protocol to produce esiRNA. The protocol is based on the in vitro digestion of long dsRNA with E. coli RNaseIII (an enzyme that generates similar molecules as Dicer in eukaryotic cells). The principle of the procedure is illustrated here.
Schematic presentation of esiRNA production. Steps carried out to
obtain a pool of functional siRNAs are illustrated. An agarose gel
before (lane 1) and after (lane 2) RNaseIII digestion is shown.
The template for in vitro transcription can be generated either by
amplification of cDNA inserts from clones using primers specific for
the vector-backbone or from cDNA preparations using target-specific
primers appended with RNA polymerase promoter sequences, such as T7,
or T3 RNA polymerase.
When choosing cDNA clones or designing cDNA-specific vectors it should
be taken into account that the amplicon length is critical for the
yield of dsRNA. We observed highest yields with amplicon lengths
between 400 and 1000 bp. While shorter templates appear to require
longer RNase III digestion times, we occasionally observed annealing
problems of the two ssRNAs when using templates longer than 1000 bp.
Taq DNA polymerase and buffers (the dye in the BioTaq Red polymerase
(Bioline) is very useful to monitor transcription efficiency; see also
in vitro transcription)
Forward and reverse primers with T7 promoter sequence 5’-
CGTAATACGACTCACTATAGGG
added 5’ to a gene-specific or vector-backbone-specific primer
sequence; internal T7 promoter sites that are often present in vector
backbones can be utilized by choosing a primer to these sites.
Thermal cycler (e.g. MJ Research)
Template (e.g. clone or cDNA)
1. Mix the following reagents in a PCR tube:
2. Amplify as follows: initial denaturation step of 94°C for 3 min;
then 35 cycles of 94°C/30 sec, 60°C/30 sec and 72°C/30 sec;
final elongation step of 72°C for 5 min.
3. Analyze products by standard agarose gel electrophoresis.
PCR products can be used directly for in vitro transcription reactions without purification. We routinely use 4 µl of the PCR product directly in in vitro transcription reactions. When the BioTaq Red polymerase from Bioline was used, the in vitro transcription reaction should result in a shift in color from red to yellow.

This figure shows 4 reactions after the in vitro transcription and annealing.
The second tube (failed) is still red and indicates that the in vitro
transcription did not work well. The third tube (good) shows a nice color shift
towards orange or yellow, indicative of an efficient reaction, most likely due
to a shift in pH.
The production of dsRNA including the synthesis of the two RNA strands and their annealing is performed in a single tube. We have further streamlined this step by skipping the conventional purification steps such as removal of DNA, nucleotides and precipitation after in vitro transcription.
1. Prepare a 10 µl reaction mix at room temperature as follows:
| 10 x transcription buffer | 1.0 µl |
| 75 mM ATP, GTP, CTP, and UTP | 1.0 µl each (use 4 µl of "NTP mix") |
| PCR product | 4.0 µl (~ 0.5 µg DNA template) |
| T7 RNA polymerase enzyme mix | 1.0 µl |
2. Incubate the reaction at 37°C overnight
3. Perform annealing in a thermal cycler as follows: 90°C for 3 min,
ramp to 70°C with 0.1°C/sec, 70°C for 3 min, ramp to 50°C
with 0.1°C/sec, 50°C for 3 min, ramp to 25°C with 0.1°C/sec.
Do not freeze the long dsRNA as it tends to form aggregates that hamper the
downstream digestion.
Long dsRNA is partially digested to esiRNAs with a length of about 18-25 bp. The subsequent purification effectively removes remaining DNA template, unincorporated nucleotides and dsRNAs longer than 40 bp. We use the GST-RNaseIII fusion protein as described in Yang et al. (http://www.pnas.org/cgi/content/full/99/15/9942). The protein is easy to purify from E.coli and stable for a long time when stored at -20°C. As E.coli RNaseIII overdigestion results in fragments sizes of ca. 12-15 bp we use a limited amount of the enzyme to achieve dsRNA length of 18-30 bp. It is therefore useful to perform a pilot serial dilution experiment with each new batch of RNaseIII to determine the optimal amount of enzyme in the digestion reaction.
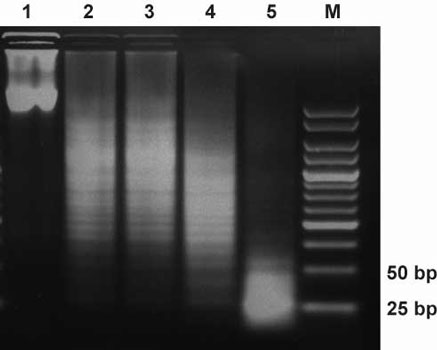
Optimization of dsRNA digestion. Lane1: dsRNA, no RNaseIII, Lane2: dsRNA,
1µg RNaseIII, Lane 3: dsRNA, 2µg RNaseIII, Lane 4: dsRNA,
3µg RNaseIII, Lane 5: 4µg RNaseIII. All reactions were incubated
at 23°C for 4 hours and then loaded on a 4% agarose gel.
Interestingly, the GST-part seems to shift the digestion products towards the desired 21-30 bp range.
1. Mix 10 µl of the in vitro transcription reaction (∼25-50 µg dsRNA)
with ∼4 µg of GST-RNase III protein in 90 µl digestion buffer
2. Incubate at 23°C for 4 h on a thermomixer at 1200 rpm.
3. Run a 2-4 µl aliquot in 4% (v/v) agarose gel along with a 25 bp DNA ladder
to check the size range of digestion products. If the size range is not appropriate
(i. e., digestion products are too long) incubate for additional 2 h at 37°C
and check again. TIP: 4% agarose gels are a bit difficult to prepare. A good
solution is to heat the aragrose in TBE in a waterbath set to 95°C. It takes
some time to dissolve the agarose that way, but it avoids the generation of
bubbles.
4. Purify the digestion products immediately as follows, or terminate the reaction
by adding 4 µl 0.5 M EDTA.
5. Prepare spin columns for purification: Add 200 µl Q-Sepharose and 500 µl
equilibration buffer to the column. Spin at 1000 g for 1 min and discard the
flow-through.
6. Again, add 500 µl equilibration buffer, spin at 1000 g for 1 min and discard
the flow-through.
7. Load all of the digested dsRNA on the column and incubate for 5 min at room
temperature.
8. Spin at 1000 g for 1 min and discard the flow-through.
9. Add 500 µl wash buffer, spin at 1000 g for 1 min and discard the flow-through.
10. Add 300 µl elution buffer, spin at 1000 g for 1 min and collect the flow-through.
11. Repeat step 10.
12. Add 500 µl isopropanol to the 600 µl eluted esiRNA and vortex. Store on
ice for at least for 30 min.
13. Spin at 16,000 g for 15 min at 4°C. Discard the supernatant, and
wash the pellet twice with cold 70% (v/v) ethanol.
14. Air-dry the esiRNA pellet for 10-15 min at room temperature and dissolve
in 50 µl of 1 x TE buffer.
15. Run a 2-4 µl aliquot on a 4% (v/v) agarose gel, and measure OD260 in order
to quantify the esiRNA concentration (for practical reasons, we typically use
the same factor for quantification of esiRNA as for dsDNA).